





- Synthesis and Curing of AUT-PNIMMO with Three Functional Groups
Xiaochuan Wang, Ping Li*,†
 , Xianming Lu, Hongchang Mo, Minghui Xu, Ning Liu, and Yuanjie Shu
, Xianming Lu, Hongchang Mo, Minghui Xu, Ning Liu, and Yuanjie ShuXi’an Modern Chemistry Research Institute, Xi’an 710065, China
*School of Mechanical and Electric Engineering, Guangzhou University, Guangzhou, 510006, China- 삼관능기를 지닌 AUT-PNIMMO의 합성 및 경화
T-PNIMMO with three functional groups was synthesized by cationic ring opening polymerization of NIMMO (3-nitratomethyl-3-methyloxetane) in the presence of TMP (trimethylolpropane) catalyzed by BF3·OEt2. Then AUT-PNIMMO was synthesized by T-PNIMMO and allyl isocyanate. The polymers were characterized by FTIR, 1H NMR, 13C NMR, and their thermal stability was estimated by DSC. Moreover, tensile testing was used to evaluate the mechanical properties of polyurethane elastomer based on T-PNIMMO and toluene-2,4-diisocyanate (TDI), and isoxazoline elastomer based on AUT-PNIMMO and tetramethyl-terephthalobisnitrile oxide (TTNO). This showed an increase of tensile strength from 2.0 to 4.0 MPa and elongation at break from 150% to 500%, respectively. These results indicated that AUT-PNIMMO exhibited a good thermal stability, had satisfactory mechanical properties and was expected to be used in the composite solid propellant and polymer bonded explosives.
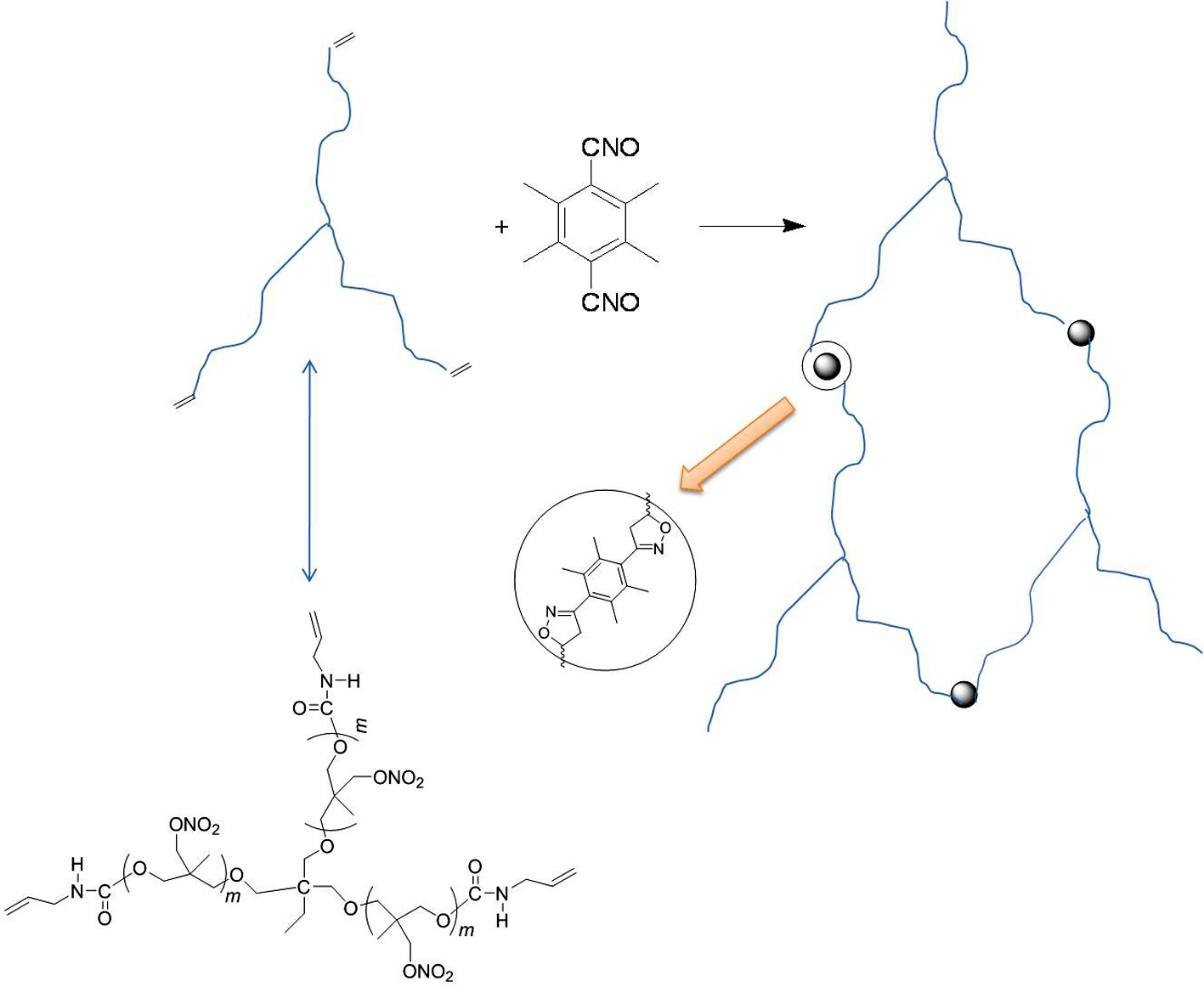
AUT-PNIMMO with three functional groups was synthesized by T-PNIMMO and allyl isocyanate. The curing of AUT-PNIMMO depends on –CNO groups react with C=C bonds to form the isoxazoline. This new curing system makes the curing temperature to room temperature and excellent improvement of the physical properties of the resulting elastomer.
Keywords: energetic binder, 3-nitratomethyl-3-methyloxetane, polymerization, isoxazoline elastomer
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (51373159).
Hydroxyl-terminated polybutadiene (HTPB) has been used in composite solid propellant and polymer bonded explosives for a very long time for binding the oxidizer, metallic fuel, and other additives.1-4 In recent years, the replacement of HTPB by energetic polymeric binders such as glycidyl azide polymer (GAP) and poly(3-nitratomethyl-3-methyloxetane) (polyNIMMO) to develop advanced rocket propellants is a new trend in the field of energetic materials. They are usually cross-linked by the curing agent of isocyanate to form polyurethanes.5-9 However, the reaction of hydroxyl terminated prepolymers with the isocyanates suffers from the humidity sensitivity in the curing process.10-13 Furthermore, the curing of isocyanate system needs high temperature.
The curing system with 1,3-dipolar cycloaddition of nitrile oxide is a different one which can decrease the curing temperature in comparison with isocyanate system.14-17 Nitrile oxides are organic compounds which contain –CNO bound directly to carbon atom.18-21 Sterically hindered bifunctional nitrile oxides are definitely stable at room temperature. The –CNO groups react with C=C bonds to form the isoxazoline. Therefore, tetramethyl-terephthalobisnitrile oxide (TTNO) can be used as a room-temperature curing agent for curing unsaturated polymers.22-26
In this study, AUT-PNIMMO was synthesized by T-PNIMMO and allyl isocyanate. For there are three C=C bonds in each AUT-PNIMMO molecule, the 1,3-dipolar cycloaddition reaction between nitrile oxide and C=C groups can be used to cure AUT-PNIMMO. T-PNIMMO with three functional groups was synthesized by cationic ring opening polymerization of 3-nitratomethyl-3-methyloxetane (NIMMO) in the presence of trimethylolpropane (TMP) catalyzed by BF3·OEt2. The structures of these compounds were confirmed by FTIR, 1H NMR, 13C NMR, and their thermal stability was estimated by DSC. The mechanical properties of isoxazoline elastomer based on AUT-PNIMMO and TTNO was compared with those of polyurethane elastomer based on T-PNIMMO and TDI.
Materials and Methods. 3-hydroxy-3-methyl-oxetane (HMMO), trimethylolpropane (TMP), toluene-2,4-diisocyanate (TDI) were purchased from J&K Scientific Ltd. (Shanghai). BF3·OEt2, CH2Cl2, Na2CO3, MgSO4 were supplied by Chengdu Kelong Chemical Reagents Company. Allyl isocyanate, HNO3 and acetic anhydride were purchased from Energy Chemical (Shanghai).
FTIR spectra were measured with a Bruker Tensor 27 instrument (KBr pellet) with a resolution of 4 cm-1 in range of 400-4000 cm-1. 1H NMR and 13C NMR spectra were recorded with a Bruker 500 MHz instrument using CDCl3 as solvent. DSC conducted with a TA Instruments DSC Q1000 was used to thermally characterize the samples using a heating/cooling rate of 10 ℃/min. According to GB/T 528-2009, the elastomer was cut out dumbbell-shaped samples for tensile test. The test was carried out on an Instron 4505 computer-controlled electronic testing machine at a rate of 100 mm/min.
Synthesis of 3-Nitratomethyl-3-Methyloxetane (NIMMO).27 HNO3 (20 mL, 0.476 mol) was dropwise added into a stirring mixture of CH2Cl2 (100 mL) and acetic anhydride (47.35 g, 0.463 mol) at 10 ℃. After stirring for an additional 30 min at 10 ℃, the reaction mixture was cooled to -4 ℃. Then 3-hydroxy-3-methyloxetane (HMMO, 35 g, 0.343 mol) was added dropwise for 2 h to the stirring solution. The mixture was stirred for another 10 min. And then, the reaction was halted by the addition of aqueous solution of Na2CO3. The organic phase was separated and washed with distilled water, and then evaporation of CH2Cl2 gave colorless oil (40.22 g, 79.7%) (Scheme 1).
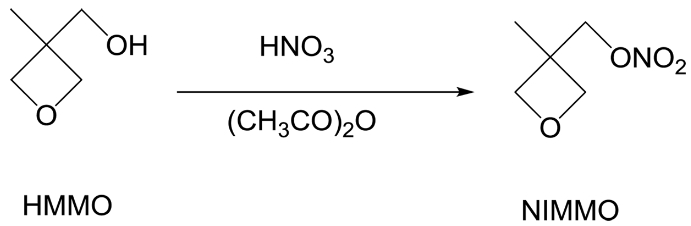
Scheme 1. Synthesis route of 3-nitratomethyl-3-methyloxetane.
Synthesis of T-PNIMMO. TMP (1.3 g, 0.01 mol) was dissolved in CH2Cl2 (20 mL) in a three-necked round bottomed flask fitted with a thermometer, BF3·OEt2 (1.4 g, 0.01 mol) was then added into the flask and stirred for 30 min at 25 ℃. NIMMO (22 g, 0.15 mol) was added dropwise for 2 h. After addition of the monomers, the reaction was left to react for another 48 h. And then, the reaction was halted by the addition of aqueous solution of Na2CO3. The organic phase was separated and washed with distilled water, and then evaporation of CH2Cl2 gave yellowish T-PNIMMO (22.43 g, 96.7%) (Scheme 2).
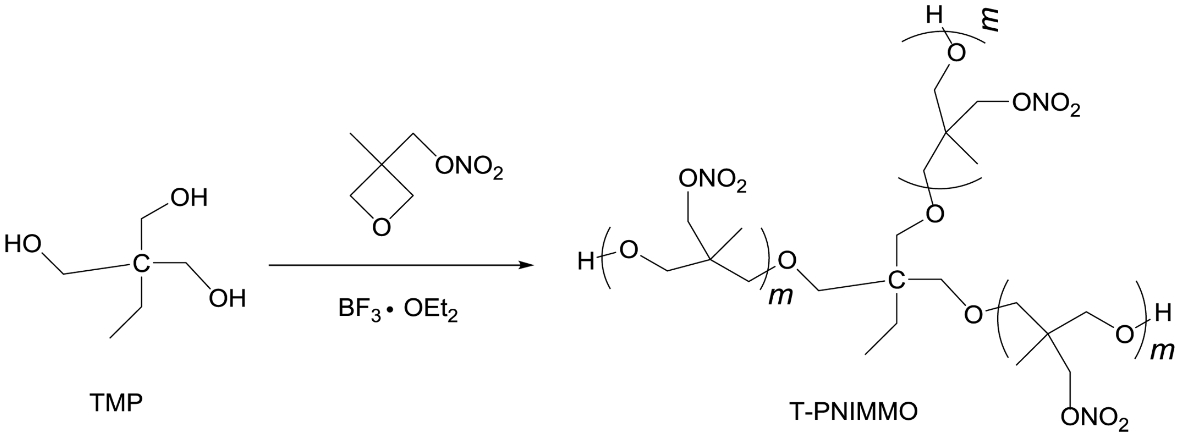
Scheme 2. Synthesis route of T-PNIMMO.
Scheme 3 shows the synthesis route of T-PNIMMO via cationic polymerization.28
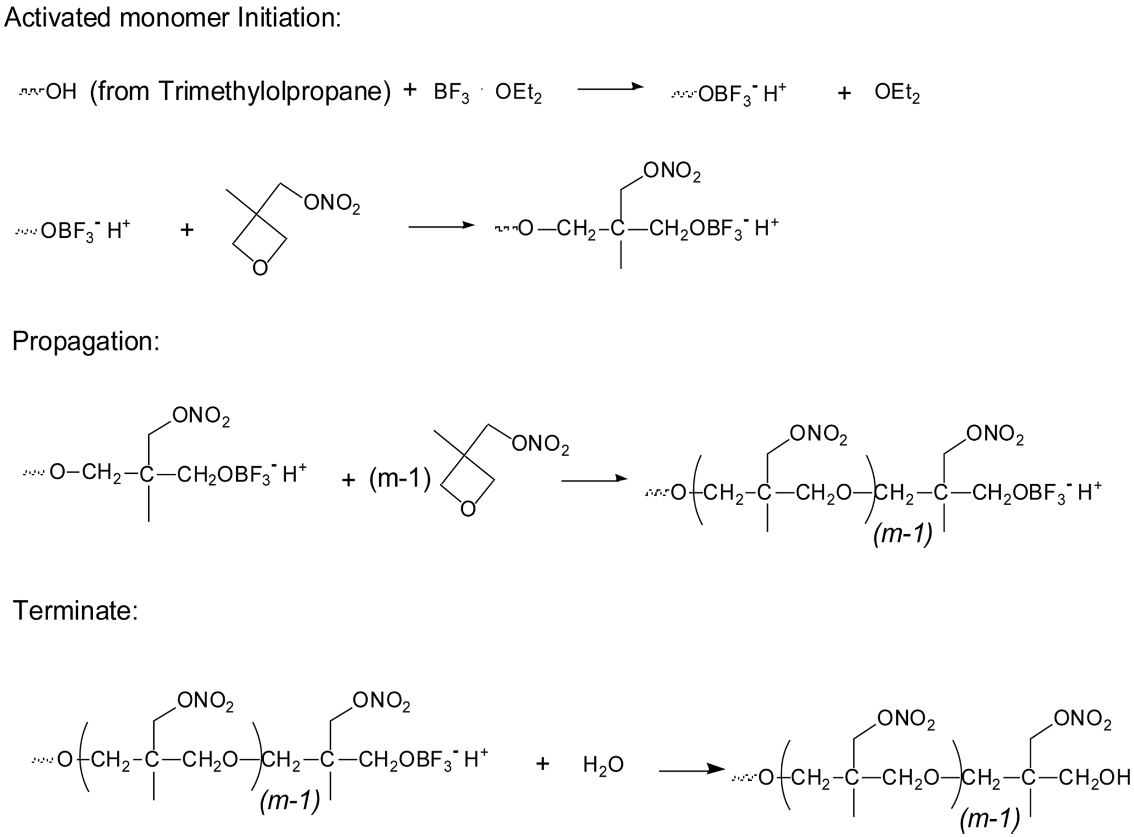
Scheme 3. Synthesis route of T-PNIMMO via cationic polymerization.
Preparation of Polyurethane Elastomer Based on T-PNIMMO and TDI.29 Polyurethane elastomer based on T-PNIMMO and TDI was prepared via mixing T-PNIMMO and TDI at a NCO/OH ratio of 1.1. A typical synthesis procedure was as follows: T-PNIMMO was mixed with TDI and degassed at rotary evaporator for 30 min. The mixture was then cast into a Teflon mold with a thickness of approximately 2 mm and left to react 7 days at 65 ℃. The obtained polyurethane elastomer was cut into dumbbell-shaped specimen to measure the mechanical properties.
Synthesis of AUT-PNIMMO. T-PNIMMO (22 g, 0.01 mol) was added in a three-necked round bottomed flask fitted with a thermometer. Allyl isocyanate (2.49 g, 0.03 mol) was added dropwise for 10 min at 50 ℃. After addition of allyl isocyanate, the reaction was left to react for 12 h at 75 ℃. And then, yellowish AUT-PNIMMO (24.49 g, 100%) was obtained (Scheme 4).
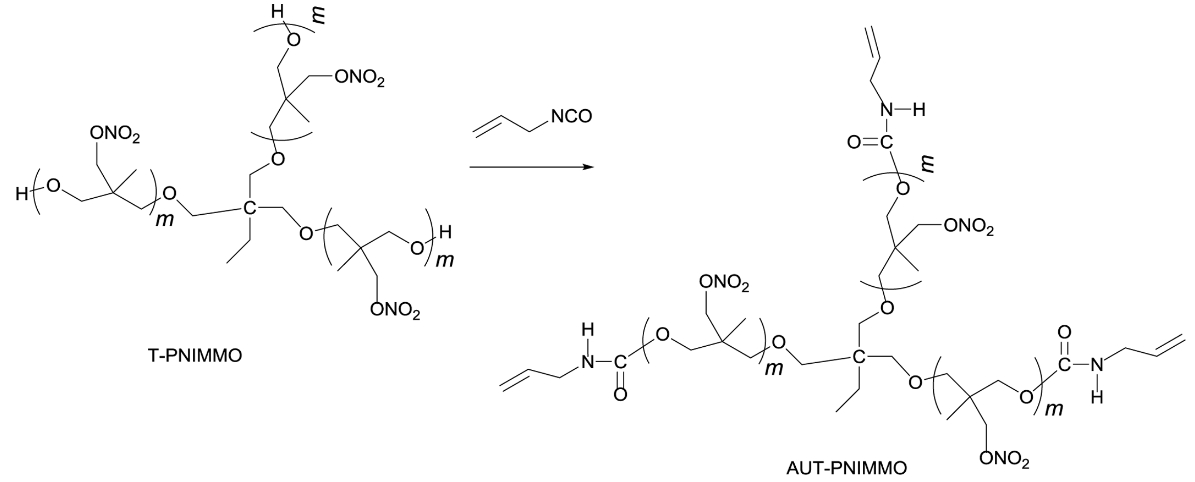
Scheme 4. Synthesis route of AUT-PNIMMO.
Preparation of Isoxazoline Elastomer Based on AUT-PNIMMO and TTNO. The TTNO was synthesized with four steps in total yield of 72%. The synthetic scheme was shown as the following formula.30 After recrystallization from methanol-methylene chloride, the TTNO was obtained analytically pure. 1H NMR (500 MHz, CDCl3), δ 2.41 (s, 12H); 13C NMR (125 MHz, CDCl3), δ 138.1, 117.5, 18.6. Anal. Calcd for C12H12N2O2: C, 66.65; H, 5.59; N, 12.96. Found: C, 67.21; H, 5.76; N, 11.90 (Scheme 5).
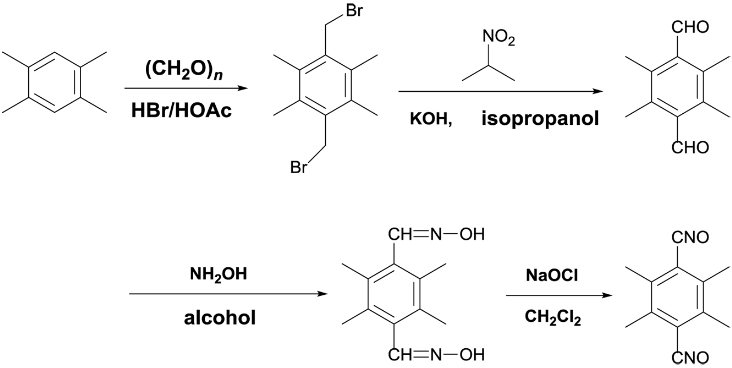
Scheme 5. Synthesis route of TTNO.
Preparation of isoxazoline elastomer based on AUT-PNIMMO and TTNO was conducted via mixing AUT-PNIMMO and TTNO at a CNO/C=C ratio of 1. A typical synthesis procedure was as follows: TTNO was mixed with CH2Cl2 to achieve a clear solution. The solution was mixed with AUT-PNIMMO for 5 h. The mixture was then cast into a petrie dish and left to react 7 days at 25 ℃. The obtained isoxazoline elastomer was cut into dumbbell-shaped specimen to measure the mechanical properties.
Structure of T-PNIMMO. Figure 1 shows the FTIR spectra of NIMMO and T-PNIMMO. In the FTIR spectrum of the NIMMO, the symmetric and unsymmetric stretching vibration of -NO2 was observed at 1630 and 1281 cm-1. The observed peak at 2886 cm-1 is attributed to stretching vibration of C-H. The FTIR spectrum of T-PNIMMO shows same characteristic peaks of NIMMO at 2886, 1630, and 1281 cm-1, the peak at 1112 cm-1 is attributed to stretching vibration of C-O in the chain.31,32 Moreover, the absorbance peak at 3444 cm-1 is attributed to O-H in T-PNIMMO.
The structures of T-PNIMMO were characterized by 1H NMR and 13C NMR as presented in Figure 2. As shown in Figure 2(A), the signals observed at 0.96-1.00 ppm were attributed to the methyl protons (denoted c) of the side chain. The signals at 3.25-3.37 ppm were due to the methylene protons (denoted b) of the main chain. The signals at 4.31-4.49 ppm were attributed to the methylene protons (denoted a) of the side chain.33 The polymerization reaction was also confirmed by 13C NMR spectrum. As shown in Figure 2(B), the resonances of methyl carbons (denoted a, b) in side chain were presented at 7.5 and 16.9 ppm, respectively. The resonances of methylene carbons (denoted c) in side chain were presented at 17.3 ppm. The resonances of quaternary carbons (denoted d, e) in the main chain were presented at 40.4 and 66.4 ppm, respectively. Meanwhile, the resonances of methylene carbons (denoted f, g and h) in the main chain were presented at 71.5, 73.7 and 74.9 ppm, respectively. The resonances of methylene carbons in the side chain were presented at 75.8 ppm (denoted i).
Thermal Properties of T-PNIMMO. Figure 3 shows the DSC glass transition temperature (Tg) curve of T-PNIMMO. The T-PNIMMO shows very low glass transition temperature (-42 ℃). The Tg value is much lower than polyNIMMO (-30 ℃).34 Furthermore, T-PNIMMO has a single Tg. The single Tg of T-PNIMMO indicates that it is a single polymer.
Figure 4 shows the DSC curve of T-PNIMMO. The degradation of polyNIMMO initiated via the breakage of nitrate ester groups at around 215 ℃.35 As can be seen, an exothermic decomposition peak is observed around 217 ℃ corresponding to degradation of –ONO2 energetic segment.
Structure of AUT-PNIMMO. Figure 5 shows the FTIR spectra of T-PNIMMO and AUT-PNIMMO. In the FTIR spectrum of the AUT-PNIMMO, the stretching vibration of N-H was observed at 3443 cm-1. The in-plane bending vibration of N-H was observed at 1518 cm-1. The peak at 1727 cm-1 is attributed to stretching vibration of C=O.36,37 The FTIR spectrum of AUT-PNIMMO shows same characteristic peaks of T-PNIMMO at 2887, 1632, and 1281 cm-1.
The structures of AUT-PNIMMO were characterized by 1H NMR and 13C NMR as presented in Figure 6. As shown in Figure 6(A), the signals observed at 0.96-1.00 ppm were attributed to the methyl protons (denoted c) of the side chain. The signals at 3.25-3.28 ppm were due to the methylene protons (denoted b) of the main chain. The signals at 4.39 ppm were attributed to the methylene protons (denoted a) of the side chain. The signals at 5.85, 5.24, and 5.13 ppm were due to the alkenyl protons (denoted d, e and f) of the main chain. The signals at 4.01 and 3.80 ppm were due to the methylene protons (denoted g, h) of the main chain. The reaction was also confirmed by 13C NMR spectrum. As shown in Figure 6(B), the resonances of carbons are similar with T-PNIMMO. The difference is shown as follows. The resonances of alkenyl carbons (denoted j, k) in the main chain were presented at 116.3 and 134.3 ppm. The resonances of carbonyl carbons (denoted l) in the main chain were presented at 155.9 ppm. The resonances of methylene carbons (denoted m) in the main chain were presented at 43.5 ppm.38
Figure 7 shows the GPC curves of T-PNIMMO and AUT-PNIMMO. It was observed that the synthesized polymer T-PNIMMO had a relatively narrow molecular weight distribution (PDI=Mw/Mn=1.42) and Mw=2900 g/mol. The Mw of the AUT-PNIMMO increased from 2900 to 3100 g/mol (PDI=Mw/Mn=1.38) after reaction. The GPC result was lower than the theoretical molecular weight of AUT-PNIMMO (around 3500 g/mol), it may be attributed to the unexpected transfer and termination processes involved in the polymerization. These results confirmed the successful synthesis of the polymer T-PNIMMO and AUT-PNIMMO.
Thermal Properties of AUT-PNIMMO. Figure 8 shows the Tg curve of AUT-PNIMMO. The Tg value (-26 ℃) of AUT-PNIMMO is higher than T-PNIMMO (-42 ℃). Furthermore, AUT-PNIMMO has a single Tg. The single Tg of AUT-PNIMMO indicates that it is a single polymer.39
Figure 9 shows the DSC curve of AUT-PNIMMO. As can be seen, an exothermic decomposition peak is observed around 217 ℃ corresponding to degradation of –ONO2 energetic segment. This is similar with the DSC curve of T-PNIMMO and indicates that AUT-PNIMMO has thermal stability.
Thermal Property of Isoxazoline Elastomer. The thermal stability of energetic materials plays an important role in the application of elastomer. Hence, that is important to research the thermal decomposition behavior for isoxazoline elastomer based on AUT-PNIMMO and TTNO. In order to investigate the dependence of elastomer structure to thermal behavior of elastomer, DSC measurement was performed. Figure 10 shows the DSC curve of isoxazoline elastomer based on AUT-PNIMMO and TTNO. An exothermic decomposition peak is observed around 220 ℃ corresponding to degradation of polyNIMMO energetic segment.
Residual solvent in the elastomer is expected to affect their physical properties. The TGA curve is used to prove that there is no residual solvent in the isoxazoline elastomer obtained after curing. The TGA curve of the isoxazoline elastomer is shown in Figure 11. The TGA thermogram displays two distinct regions of weight loss. The first weight loss temperature occurred at 220 ℃ with a sharp weight loss of around 40% with respect to the total, corresponding to the decomposition of –ONO2 energetic segment. The second main weight loss occurred at 240-400 ℃ with a gradual weight loss was due to the main-chain thermal decomposition of the isoxazoline elastomer.
The Mechanical Properties of Isoxazoline Elastomer and Polyurethane Elastomer. Tensile testing evaluated the ultimate mechanical properties of polyurethane elastomer based on T-PNIMMO and TDI and isoxazoline elastomer based on AUT-PNIMMO and TTNO (Table 1). The tensile strength and elongation at break of polyurethane elastomer were 2.0 MPa and 150%, respectively. The tensile strength and elongation at break of isoxazoline elastomer were 4.0 MPa and 500%, respectively.
The FTIR spectra of the mixture of AUT-PNIMMO and TTNO, isoxazoline elastomer and AUT-PNIMMO are shown in Figure 12. Comparison of the spectra shows that the disappearance of strong absorption peak (2291 cm-1) of –CNO indicates the completion of 1,3-dipolar cycloaddition reaction. There are three C=C bonds in each AUT-PNIMMO molecule and two –CNO groups in each TTNO molecule. The –CNO group of TTNO reacts with C=C bond of AUT-PNIMMO to form the isoxazoline. As a result, the FTIR absorption peak of –CNO group disappeared. These can confirm successful progress of the reaction of nitrile oxides with vinyl end groups in AUT-PNIMMO.
Figure 13 shows the formation of isoxazoline elastomer based on AUT-PNIMMO and TTNO. As can be seen, the strong hydrogen bonding interaction existing in urethane and isoxazoline has remarkable effect on the mechanical properties of the isoxazoline elastomer.40 As a result, the isoxazoline elastomer based on AUT-PNIMMO and TTNO has showed improved mechanical properties than polyurethane elastomer based on T-PNIMMO and TDI.

|
Figure 1 FTIR spectra of NIMMO and T-PNIMMO. |

|
Figure 2 (A) 1H NMR; (B) 13C NMR of T-PNIMMO. |

|
Figure 3 DSC glass transition temperature (Tg) curve of T-PNIMMO. |

|
Figure 4 The DSC curve of T-PNIMMO. |

|
Figure 5 FTIR spectra of T-PNIMMO and AUT-PNIMMO. |

|
Figure 6 (A) 1H NMR; (B) 13C NMR of AUT-PNIMMO. |

|
Figure 7 GPC curves of T-PNIMMO and AUT-PNIMMO. |

|
Figure 8 DSC glass transition temperature (Tg) curve of AUT-PNIMMO. |

|
Figure 9 DSC curve of AUT-PNIMMO. |

|
Figure 10 DSC curve of isoxazoline elastomer. |

|
Figure 11 TGA curve of isoxazoline elastomer. |

|
Figure 12 FTIR spectra of the mixture of AUT-PNIMMO and TTNO, isoxazoline elastomer and AUT-PNIMMO. |

|
Figure 13 Formation of isoxazoline elastomer. |
|
Table 1 Mechanical Properties of Isoxazoline Elastomer and Polyurethane Elastomer |

AUT-PNIMMO was synthesized by T-PNIMMO and allyl isocyanate to produce a material for energetic binder for polymer bonded explosives and solid rocket propellant. T-PNIMMO with three functional groups was synthesized by cationic ring opening polymerization of 3-nitratomethyl-3-methyloxetane (NIMMO) in the presence of trimethylolpropane (TMP) catalyzed by BF3·OEt2. The structures of these compounds were confirmed by FTIR, 1H NMR, 13C NMR, and their thermal stability was estimated by DSC. Tensile testing evaluated the ultimate mechanical properties of polyurethane elastomer based on T-PNIMMO and TDI and isoxazoline elastomer based on AUT-PNIMMO and TTNO. That showed an increase of tensile strength from 2.0 to 4.0 MPa and elongation at break from 150% to 500%, respectively. These results indicated that AUT-PNIMMO exhibited a good thermal stability, and was expected to be used in the composite solid propellant and polymer bonded explosives.
- 1. B. S. Min and H. H. Jeong, Polym. Korea, 42, 192 (2018).
-
- 2. M. S. Khan, A. Dey, J. Athar, and A. K. Sikder, RSC Adv., 4, 32840 (2014).
-
- 3. Z. J. Zhang, N. Luo, Z. Wang, and Y. J. Luo, J. Appl. Polym. Sci., 132, 42026 (2015).
-
- 4. X. C. Wang, Y. J. Shu, X. M. Lu, H. C. Mo, and M. H. Xu, Cent. Eur. J. Energ. Mater., 15, 456 (2018).
-
- 5. R. M. Shankar, T. K. Roy, and T. Jana, J. Appl. Polym. Sci., 114, 732 (2009).
-
- 6. F. Cauty, Y. Fabignon, and C. Erades, Inter. Energ. Mater. Chem. Propul., 12, 1 (2013).
-
- 7. C. S. Pant, M. Santosh, S. Banerjee, and P. K. Khanna, Propell. Explos. Pyrotech., 38, 748 (2013).
-
- 8. S. H. Kim, S. B. Kim, and S. H. Cha, Polym. Korea, 42, 736 (2018).
-
- 9. D. Lee, K. T. Kim, Y. Jang, S. Lee, H. B. Jeon, H. Paik, B. S. Min, and W. Kim, J. Appl. Polym. Sci., 131, 4401 (2014).
-
- 10. V. Vasudevan and G. Sundararajan, Propell. Explos. Pyrotech., 24, 295 (1999).
-
- 11. M. H. Xu, Z. X. Ge, X. M. Lu, H. C. Mo, Y. P. Ji, and H. M. Hu, Polym. Int., 66, 1318 (2017).
-
- 12. T. Keicher, W. Kuglstatter, S. Eisele, T. Wetzel, and H. Krause, Propell. Explos. Pyrotech., 34, 210 (2009).
-
- 13. S. Reshmi, E. Arunan, and C. Nair, Ind. Eng. Chem. Res., 53, 16612 (2014).
-
- 14. H. Li, F. Q. Zhao, Q. Q. Yu, B. Z. Wang, and X. M. Lu, J. Appl. Polym. Sci., 133, 43341 (2016).
-
- 15. M. Badgujar, M. B. Talawar, S. N. Asthana, and P. Mahulikar, J. Hazard. Mater., 151, 289 (2008).
-
- 16. P. Yakubov, V. Tsyganov, I. Belenkii, and M. Krayushkin, Russ. Chem. B+, 40, 1078 (1991).
-
- 17. J. Park and C. Y. Lee, Polym. Korea, 42, 371 (2018).
-
- 18. I. Belenkii, P. Gromova, B. V. Lichitskii, and M. Krayushkin, Russ. Chem. B+, 46, 101 (1997).
-
- 19. Y. Li and B. Cheng, J. Polym. Sci., Part A: Polym. Chem., 51, 1645 (2013).
- 20. S. A. Kotelnikov, V. S. Sukhinin, and A. S. Ermilov, Russ. J. Appl. Chem.+, 75, 477 (2002).
-
- 21. Y. Koyama, M. Yonekawa, and T. Takata, Chem. Lett., 37, 918 (2008).
-
- 22. B. S. Huffman, R. A. Schultz, and P. J. Schlom, Polym. Bull., 47, 159 (2001).
-
- 23. J. J. Tegeler and C. J. Diamond, J. Heterocyclic Chem., 24, 697 (1987).
-
- 24. H. Choe, P. T. Trang, J. Y. Lee, M. Latif, H. Park, Y. K. Kang, and J. Lee, J. Org. Chem., 81, 2612 (2016).
-
- 25. C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat, and P. Kittakoop, Angew. Chem. Int. Ed., 55, 3997 (2016).
-
- 26. Z. X. Yu and K. N. Houk, J. Am.Chem. Soc., 125, 13825 (2003).
-
- 27. P. Golding, R. W. Millar, N. C. Paul, and D. H. Richards, Tetrahedron Lett., 32, 4985 (1991).
-
- 28. A. U. Francis, S. Venkatachalam, M. Kanakavel, V. Ravindran, and K. N. Ninan, Eur. Polym. J., 39, 831 (2013).
- 29. D. Bhowmik, V. S. Sadavarte, S. M. Pande, and B. S. Saraswat, Cent. Eur. J. Energ. Mater., 12, 145 (2015).
- 30. X. C. Wang, Y. J. Shu, Y. L. Lei, Y. Q. Fan, X. M. Lu, and Y. Shu, Fine Chem., 35, 695 (2018).
- 31. H. J. Desai, A. V. Cunliffe, T. Lewis, R. W. Millar, N. C. Paul, M. J. Stewart, and A. J. Amass, Polymer, 37, 3471 (1996).
-
- 32. H. J. Desai, A. V. Cunliffe, J. Hamid, P. J. Honey, M. J. Stewart, and A. J. Amass, Polymer, 37, 3461 (1996).
-
- 33. M. H. Xu, Z. X. Ge, X. M. Lu, H. C. Mo, Y. P. Ji, and H. M. Hu, RSC Adv., 7, 47271 (2017).
-
- 34. H. C. Mo, X. X. Gan, Y. Xing, and N. Li, Chinese J. Explos. Propel., 29, 58 (2006).
- 35. Q. L. Yan, A. Cohen, A. K. Chinnam, N. Petrutik, A. Shlomovich, L. Burstein, and M. Gozin, J. Mater. Chem. A, 4, 18401 (2016).
-
- 36. F. Abrishami, N. Zohari, and V. Zeynali, Propell. Explos. Pyrotech., 42, 1 (2017).
- 37. M. Ghorbani and Y. Bayat, Polym. Sci. Ser. B+, 57, 654 (2015).
-
- 38. Y. M. Mohan, M. P. Raju, and K. M. Raju, Int. J. Polym. Mater. Po., 54, 651 (2005).
-
- 39. S. H. Jeong, Y. S. Lee, and K. H. Yoon, Polym. Korea, 43, 10 (2019).
-
- 40. H. Li, Q. Q. Yu, F. Q. Zhao, B. Z. Wang, and N. Li, J. Appl. Polym. Sci., 134, 45178 (2017).

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2024 Impact Factor : 0.6
- Indexed in SCIE
 This Article
This Article
-
2019; 43(4): 503-511
Published online Jul 25, 2019
- 10.7317/pk.2019.43.4.503
- Received on Nov 17, 2018
- Revised on Mar 18, 2019
- Accepted on May 3, 2019
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Xiaochuan Wang, Ping Li
-
*School of Mechanical and Electric Engineering, Guangzhou University, Guangzhou, 510006, China
- E-mail: leeping0521@126.com
- ORCID:
0000-0002-6445-7910
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr