






- Synthesis and Characterization of Cellulose Acetate-g-Poly(L-lactic acid)
Hyo Sung Choi, Ga Yeun Kim, and Young Ho Kim†

Department of Organic Materials and Fiber Engineering, Soongsil University, Seoul 06978, Korea
- 셀룰로오스 아세테이트-g-폴리(L-락트산)의 합성과 분석
최효성 · 김가연 · 김영호†
숭실대학교 유기신소재·파이버공학과
Although blends of cellulose
acetate (CA) and poly(L-lactic acid) (PLA) would show good
biodegradability, the mechanical property deterioration of the blends due to
the immiscibility of the two polymers refrains their using. In this study, CA-g-PLA
which could be used as a compatibilizer of the blends was synthesized via a
ring-opening polymerization of L-lactide. CA-g-PLAs with various
graft yields were synthesized by varying the weight ratio of L-lactide
to CA and reaction time. Then the properties of the CA-g-PLA such as
graft yield and degree of the substitution were characterized by using TGA,
GPC, FTIR, and 1H NMR spectroscopy. Graft yields of the CA-g-PLA
copolymers were determined by both TGA and 1H NMR analysis. The
graft yield of CA-g-PLA increased with increasing the weight ratio of L-lactide
to CA up to 10:1 and the grafting time up to 16 hours at 130 °C. All the
analysis results indicated that CA-g-PLA exhibits copolymer properties
instead of simple mixture properties of CA and PLA
셀룰로오스 아세테이트(CA)를
폴리(L-락트산) (PLA)과 블렌드시키면 생분해성이
향상될 것으로 기대되지만 두 고분자 사이에 혼화성이 없어 물성 저하가 심해 사용하기 곤란하다. 본 연구에서는
이들 블렌드계의 상용화제로 사용될 수 있는 CA-g-PLA를 L-락타이드의 개환중합을 이용하여 합성하였다. CA에 대한 L-락타이드의
무게비와 반응 시간을 달리하여 여러가지 그래프트율을 갖는 CA-g-PLA를 얻고, 이들의 그래프트율, 치환도 등의 특성을 열중량분석기(TGA), 젤투과 크로마토그래피, FTIR, 1H NMR 분광분석기를
사용하여 분석하였다. TGA와 1H NMR 분석에 의한 그래프트율이 서로 일치하였으며, 그래프트 반응
시 사용한 CA에 대한 L-락타이드의 비율이 커지고 반응 시간이 길어질수록 그래프트율이 크게 나타났다. 모든
분석 결과에서 CA-g-PLA는 CA와 PLA이 단순 혼합이 아닌 공중합체로서의 특성을 나타내었다
Cellulose acetate-g-poly(L-lactic acid) (CA-g-PLA)
was synthesized via a ring-opening polymerization of L-lactide in order
to use it as a compatibilizer for CA/PLA blends. CA-g-PLAs with various
graft yields were obtained by varying the weight ratio of L-lactide to
CA and reaction conditions. Graft yields of the copolymers were determined by
both TGA and 1H NMR analysis.
Keywords: cellulose acetate, poly(L-lactic acid), graft yield, thermogravimetric analysis (TGA), proton nuclear magnetic resonance (1H NMR).
이 연구는 산업통상자원부 및 산업기술평가관리원(KEIT)의 연구비 지원으로 수행되었으며(과제번호 10052095), 이에 감사드립니다.
석유계 고분자에서
나타나는 여러 가지 환경 문제들 때문에 이들을 대체할 바이오 플라스틱에 대한 연구가 활발히 이루어지고 있다. 바이오 플라스틱은 생분해성 플라스틱과
재생가능한 바이오매스 플라스틱을 포함하는데, 보통 재생가능한 원재료로 만들어지는 플라스틱을 말한다. 이는 자연계에서 쉽게 얻을 수 있을 뿐만 아니라
폐기 과정에서 생분해가 가능하여 환경오염 물질의 유발이 적다는 장점이 있다.
여러 바이오 플라스틱 중에서 자연에 가장 풍부한 셀룰로오스로부터 얻어지는 유도체인 셀룰로오스
아세테이트(CA)와 옥수수 전분으로부터 제조할 수 있는 폴리(L-락트산)(PLA)이 있다. CA는 포장 용기, 섬유, 담배 필터,
흡수체 등에 주로 이용되며, 투명성, 내정전기성, 생체적합성, 우수한 기계적 물성 등의 장점을 나타낸다.1,2 그러나
CA는 유리전이온도가 약 180 °C이고, 용융온도가 약 230 °C로 두 온도가 상당히 높고 열안정성이 낮아 용융상태에서
장시간 유지하여 제품화하기 곤란하다. 또한 CA는 아세틸 치환도가 2.2 이하일 경우에는 토양이나 해양에서 생분해성을 나타내지만, 아세틸 치환도가
그 이상인 경우에는 생분해성이 저하된다는 단점이 있다.1,3,4 이처럼 가공이 어려운 CA의 문제점을 극복하기 위해
트리아세틴, 에틸렌 글리콜, 프로필렌 글리콜, 폴리(카프로락톤 트리올) 등과 같은 가소제를 사용하여 가소화시키거나, 다른 고분자와 블렌딩 또는
그래프팅을 하여 이를 개선하는 연구가 진행되고 있다.3,4
한편, PLA는 대표적인
바이오 플라스틱으로 CA와 같은 에스터계 고분자이다. 주로 L-락트산을 축합중합하거나 L-락타이드를 개환중합하여 얻을
수 있다.5,6 PLA는 생분해성을 가지며 유리전이온도가 약 50 °C로 상대적으로 CA보다 낮아 CA와 함께 사용한다면
가공이 쉬워지고 생분해성 조절이 가능할 것이라 기대된다. 그러나 PLA와 CA 두 고분자 사이에는 혼화성(miscibility)이 없어 서로 블렌딩을
할 경우 물성 저하가 심해 함께 사용하는 데 어려움이 있다.7
두 가지 이상의 고분자를
함께 사용하여 단점을 보완하고 장점을 취하기 위해 이들을 블렌딩을 할 경우, 혼합하는 고분자들 사이의 혼화성이 중요하게 된다. 두 고분자 사이에
혼화성이 없는 경우에는 물성 저하가 매우 심해 사용에 어려움이 있다. 이 때 혼화성이 없는 두 고분자 사이의 상용성(compatibility)을
향상시켜주는 방법으로 상용화제(compati-bilizer)를 첨가해 주는 방법이 있다. 예를 들어 Ogata 등은7
티타늄 테트라이소프로파놀레이트(TP)를 CA와 PLA의 상용화제로 하여 균일한 상의 CA/PLA 블렌드 필름을 얻고, 가수분해성을 향상시키는 논문을
발표한 바 있다. 그러나 TP는 독성이 있어 인체에 해롭고 CA와 PLA의 가장 큰 장점인 인체적합성과 생분해성을 해치기 때문에 상업적으로 적용시키기
곤란하다.
본 연구에서는 이러한
점을 고려하여 PLA/CA 블렌드의 상용성을 개선시키는데 사용될 수 있는 상용화제를 합성하고자 하였다. PLA와 CA의 특성을 모두 나타내는 공중합체를
합성하기 위하여 CA를 주사슬로 하고 CA의 히드록시기에 PLA를 그래프팅시켜 PLA를 가지사슬로 갖는 CA-g-PLA 공중합체를
합성하였다. PLA를 CA에 그래프팅시키기 위해서 PLA의 단량체인 L-락트산과 L-락타이드를 사용하는 방법이 있는데
본 연구에서는 L-락타이드를 개환중합하는 방법을 이용하였다. L-락타이드는 안정한 육각환 물질로 L-락트산을
단량체로 하는 축합중합과 비교하였을 때 반응 효율이 좋고, 고분자량의 PLA를 얻을 수 있을 뿐만 아니라 분자량을 제어하기 쉽다는 장점이 있다.5,6
CA-g-PLA를
합성할 때 용융 공정이나 용액 공정을 사용할 수 있다. 용융법은 단량체를 용융시킨 상태에서 주로 용융압출기(extruder)를 사용하여 진행된다.
예를 들어 Quintana 등은8 brabender internal mixer를 사용하여 CA와 락타이드를 235 °C에서 45분간 용융압출시킴으로써 반응용융공정(reactive
melt processing)에 의해 시료를 얻고, 이를 다시 클로로포름에 용해시킨 후 메탄올/톨루엔 혼합용매에서 침전시켜 미반응 L-락타이드와
호모 PLA를 제거하는 방법을 사용하였다. 이 경우 CA 주사슬에 PLA를 그래프팅시키기 위해서는 CA와 L-락타이드가 모두 용융된
상태에서 반응이 진행되어야 보다 균일한 반응이 이루어지지만, CA의 열안정성이 낮아 용융상태를 길게 하면서 CA-g-PLA를 합성하기에는
어려움이 있다. 또한 이러한 용융공정을
이용하는 경우에는 균일한 혼합을 위해서 많은 약제가 필요하며 촉매와 같이 소량인 경우에는 균일하게 혼합하기 곤란하다. 뿐만 아니라 반응후 미반응
L-락타이드나 호모 PLA를 제거하기 위하여 얻어진 반응물을 다시 용매에 용해시킨 후 비용매에서 침전시켜 CA-g-PLA를
얻어야 한다.8,9 따라서 용액법보다 합성 방법 자체는 간단하더라도 합성
후의 정제 과정이 복잡하고 용액법과 마찬가지로 용매를 사용해야 한다는 문제점이 있다. 이를 고려하여 본 연구에서는 CA를 열분해시키지
않으면서 보다 균일한 합성과 반응도 조절을 위해 용액 상태에서 CA-g-PLA를 합성하는 방법을 사용하였다. 이 경우 열분해를 방지할
뿐만 아니라 반응시간이나 온도와 같은 반응조건을 쉽게 변경할 수 있다는 장점이 있다.
일반적으로 L-락타이드의
개시는 히드록시기에서 활성화되기 때문에 히드록시기를 갖는 디올을 개시제로 하는 방법이 오래 전부터 연구되었다.10,11 그러나
본 연구에서는 CA에 존재하는 히드록시기가 L-락타이드의 개시점으로 활성화되기 때문에 추가적인 개시제를 사용하지 않았다. 촉매로는
L-락타이드 개환중합에서 주로 이용되는 Sn(II) 2-에틸헥사노에이트를 사용하였다.8-10 Sn(II) 2-에틸헥사노에이트는
FDA 승인을 받은 만큼 인체친화성을 가지기 때문에 CA-g-PLA의 합성 과정이 보다 인체 및 환경 친화적일 것이라 판단된다.11,12
본 연구에서는 합성한
여러 가지 CA-g-PLA 공중합체를 FTIR, 1H NMR, TGA, GPC 등을 사용하여 분석하고 그래프트율을
구하였다. 이 CA-g-PLA가 실제 CA/PLA 블렌드의 상용화제로 작용하는지에 대해서는 다른 논문에 발표할 예정이다.
시료 및 시약. CA는 아세틸 치환도 2.5, 수평균 분자량
30000 g/mol인 분말상의 Sigma Aldrich사(미국)
제품을, L-락타이드는 순도 99%인 Purac사(네덜란드) 제품을 그대로 사용하였다. 디메틸설폭사이드(DMSO), 메탄올, 톨루엔,
테트라하이드로퓨란(THF)은 삼전정밀화학사 제품을, Sn(II) 2-에틸헥사노에이트는 Sigma Aldrich사 제품을, DMSO-d6는
Cambridge Isotope Laboratories 사(미국) 제품을 사용하였으며, 이들은 모두 정제하지 않고 그대로
사용하였다.
호모 PLA의 합성. CA-g-PLA 공중합체 합성에 앞서 L-락타이드의
개환중합 반응 확인과 비교를 위해 필요한 호모 PLA 고분자를 얻기 위하여 L-락타이드를 다이머로 하여 PLA를 합성하였다. 이를
위하여 교반기, 냉각기, 온도계, 온도 조절기, 질소 유입구, 적하 깔대기가 장착된 4구 플라스크에서 합성을 진행하였다. 플라스크에 DMSO
30 mL를 넣고 질소 기류 하에서 온도를 120°C까지 승온시킨
뒤 50 g의 L-락타이드를 투입하여 완전히 용해시켜
주었다. 균일한 하나의 상이 된 것을 확인한 뒤, 130 °C까지 승온시키고 촉매이자 개시제인 Sn(II) 2-에틸헥사노에이트를 L-락타이드에 대해
0.2 wt% 첨가한 후 8시간 동안 교반시키면서 반응시켰다.
반응 후 내용물을 다량의 메탄올에 침전시키고 메탄올로 수차례 수세하여 미반응 L-락타이드를 제거하였다. 이들을 감압여과한 후 60 °C의 진공오븐에서 48시간 이상 건조하여 호모
PLA를 얻었다. 이 호모 PLA는 CA-g-PLA의 특성을 분석할 때 비교를 위해서 사용하였다.
CA-g-PLA의 합성. 여러 그래프트율을 갖는 CA-g-PLA 공중합체를
합성하기 위해 교반기, 냉각기, 온도계, 온도 조절기, 질소 유입구, 적하 깔대기가 장착된 4구 플라스크에서 반응을 진행하였다. 질소 기류 하에서
DMSO 100 mL를 넣고, 온도를 120 °C까지 승온시킨 뒤 CA와 L-락타이드를
1:2, 1:6, 1:10, 1:14 의 무게비로 투입하여 완전히 용해시켜 주었다. 이 때 사용한 CA의 양은 5 g으로 일정하게 하였으며, CA와 L-락타이드
비율에 따라 반응물의 전체 무게는 15 g에서 75 g까지 변하였다. CA와 L-락타이드가 완전히 용해된 것을 확인한 후, 130 °C까지 승온시키고 촉매인 Sn(II) 2-에틸헥사노에이트를 L-락타이드에 대해 0.2 wt% 첨가한 후 8, 12, 16, 24시간 동안 교반시키면서
반응시켰다. 반응물을 다량의 메탄올/톨루엔 혼합용매(8:2, v/v)에 침전시키고 여과한 후, 같은 비율의 메탄올/톨루엔 혼합용매로 수차례 수세하여
호모 PLA, 미반응 L-락타이드 및 촉매를 제거하였다. 이들을 감압여과한 후 진공오븐에서 48시간 건조하여 여러 그래프트율을 가지는
CA-g-PLA를 얻었다. 각 시료의 그래프트율은 TGA 및 1H NMR 분석으로부터 구하였는데, 이에 대해서는 결과 및
토론에서 자세히 설명하기로 한다. 시료명에 그래프트율을 포함시켜야 하는 경우 CA-g(xx)-PLA로 표기하였는데, 괄호
안의 xx는 TGA 분석에 의하여 구한 % 그래프트율을 나타낸다.
각종 분석. 시료를 DMSO-d6에
용해시킨 후 Bruker사의 NMR(Advance 400 NMR)을 사용하여 스캔수 32로 하여 1H NMR 스펙트럼을 얻었으며, Bruker사의
FTIR(Vertex 70)을 사용하여 분말 상태의 시료를 ATR 법으로 600~4000 cm-1 범위에서 분해능 2 cm-1로 하면서 64회 스캔하여
FTIR 스펙트럼들을 얻었다. 얻어진 FTIR 스펙트럼들은 전체 면적을 1로 하여 표준화(nomalization)시켰다. Mettler
Toledo사의 TGA를 사용하여 질소기류 하에서 10 °C/min으로 승온시키면서 열분해 곡선을 얻었다. CA-g-PLA 시료들을 THF에 용해시킨 후, 3개의 Plgel Wmixed-c 컬럼(Agilent)을
사용하면서 Water사 2414 Refractive Index Detector를 디텍터로 하는 Waters사의 GPC 시스템을 사용하여 25 °C에서 GPC 크로마토그램을 얻었다. 이때 운반 액체는
THF이었다.
모 PLA의 중합 확인. CA-g-PLA의 여러 가지 특성을 분석하고
그래프트된 고분자들의 비교를 위해서는 호모 CA와 호모 PLA 고분자가 필요하다. 이때 PLA는 시판 제품보다는 CA-g-PLA 합성
시와 같은 조건에서 합성한 호모 PLA를 사용하는 것이 바람직하다. 이러한 비교와 함께 L-락타이드가 제대로 개환되어 호모 PLA로
중합되었는지를 먼저 확인하였다.
한편, 예비실험 결과
L-락타이드를 DMSO 용액에서 개환중합시킬 때의 반응온도는 130 °C가 적합하다고 판단하였다. 이는 120 °C에서 반응시킨 경우에는 용액의 점도 증가로부터 판단할 때 PLA가 중합되기까지 상당히 긴 시간이 소요되었고,
140 °C 이상에서 반응시키면 중합된 PLA가 노란색을 나타내었기
때문이었다. 뿐만 아니라 에스터계 고분자인 PLA는 높은 온도에서 에스터 교환 부반응에 의해 해중합이 일어나기 때문에13-16
분자량이 작아질 가능성이 크다. 변색이 되거나 너무 긴 반응 시간은 상업적 측면이나 공정적 측면에서 비효율적이기 때문에, 이러한 실험 결과를 바탕으로
하여 CA 주사슬에 PLA를 그래프팅시킬 때에도 130 °C의 반응온도가 적합하다고 판단하였다.
실험에서 설명한 방법으로
호모 PLA를 얻고, 출발 물질들과 이의 FTIR 스펙트럼을 측정하여 Figure 1에 나타내었다. L-락타이드의 스펙트럼(Figure 1(a))을
살펴보면, 1267 cm-1에서 락톤링의
-C-O-C- 비대칭 신축운동, 1099 cm-1에서 락톤링의 -C-O-C- 대칭 신축운동, 930 cm-1에서 -COO 링 밴드들이 나타났다.13,17,18 그러나
PLA 스펙트럼(Figrue 1(b))에서는 L-락타이드의 고리에서 기인한 밴드들이 사라지고, 1452 cm-1에서 -CH 굽힘운동, 1182 cm-1에서 -C-O-C- 대칭 신축운동, 1129 cm-1에서 -CH3
비대칭 굽힘운동, 1087 cm-1에서 지방족 사슬 -C-O-C-의 대칭 신축운동 밴드들이9,17,18 나타나고 있어, L-락타이드의 개환중합이 성공적으로 이루어져
PLA가 제대로 중합되었음을 확인할 수 있었다.
CA-g-PLA 공중합체의 합성 확인. L-락타이드로부터 호모 PLA를 중합시킬 때의 실험 결과를
토대로 CA-g-PLA를 합성하는 반응 온도를 130 °C로 설정하였다. 여러 가지 그래프트율을 가지는 CA-g-PLA를 얻기 위해 주사슬인 CA에
반응시키는 L-락타이드의 무게 비율과 반응 시간 등을 달리하여 합성을 진행하였다. CA에 대한 L-락타이드의 무게비를
1:1로 한 조건에서는 그래프트율이 너무 작게 나타나, 본 연구에서는 L-락타이드:CA의 최소 무게 비율을 2:1로 하고 L-락타이드
양을 이보다 많이 하면서 실험을 진행하였다.
본 연구에서 사용한
CA는 아세틸 치환도가 2.5인 것이다. 따라서 이 CA에는 AGU(anhydro glucose unit)당 히드록시기가 0.5 몰 존재하며 이
히드록시기에 L-락타이드가 그래프트되어 PLA를 형성한다. CA 주사슬에 PLA가 그래프트된 CA-g-PLA의 IR 스펙트럼은
CA와 PLA에서 기인되는 피크를 모두 나타낼 것으로 기대된다. 시료의 그래프트율을 구하는 방법은 뒤에서 설명할 예정이지만, 그래프트율이
50.10%인 CA-g(50.1)-PLA와 CA의 FTIR 스펙트럼을 얻어 Figure 1에 함께
나타내었다. CA-g-PLA의 주사슬은 CA이기 때문에 공중합체의 FTIR 스펙트럼은 CA 스펙트럼에 PLA의 피크가 더해진 형태로
나타나며, 히드록시기에 PLA가 그래프트되면서 3476 cm-1에서 나타나는 CA의 히드록시기 피크19,20
크기가 크게 감소하였음을 확인할 수 있다. CA-g(50.1)-PLA의 IR 스펙트럼을 보면, 기존 CA에 기인한 밴드들
이외에 PLA에 기인한 -CH3 굽힘운동 밴드가 1456 cm-1에서, -C-O-C- 대칭
신축운동 밴드가 1182 cm-1에서, -CH3
비대칭 굽힘운동 밴드가 1129 cm-1에서, 지방족 사슬
-C-O-C- 진동 밴드가 1087 cm-1에서13,17,18 나타나고 있어 CA 주사슬에 PLA가 그래프트된 것을
확인할 수 있다.
한편, CA-g-PLA가
합성될 때 L-락타이드의 개환으로 인해 그래프트된 PLA 뿐만 아니라 호모 PLA도 생성될 수 있다. CA-g-PLA를
CA/PLA 블렌드의 상용화제로 사용하는 용도만을 고려한다면 호모 PLA를 제거시킬 필요가 없지만, CA-g-PLA의 정확한 특성
분석을 위해서는 호모 PLA를 모두 제거시킨 순수한 공중합체가 필요하다. 본 연구에서는 PLA의 용매인 톨루엔과 CA와 PLA의 비용매인 메탄올의
혼합용매 (2:8, v/v)를 사용하여 반응물을 침전 및 수세하여 호모 PLA를 제거하였다. 얻어진 CA-g-PLA 시료에서 호모
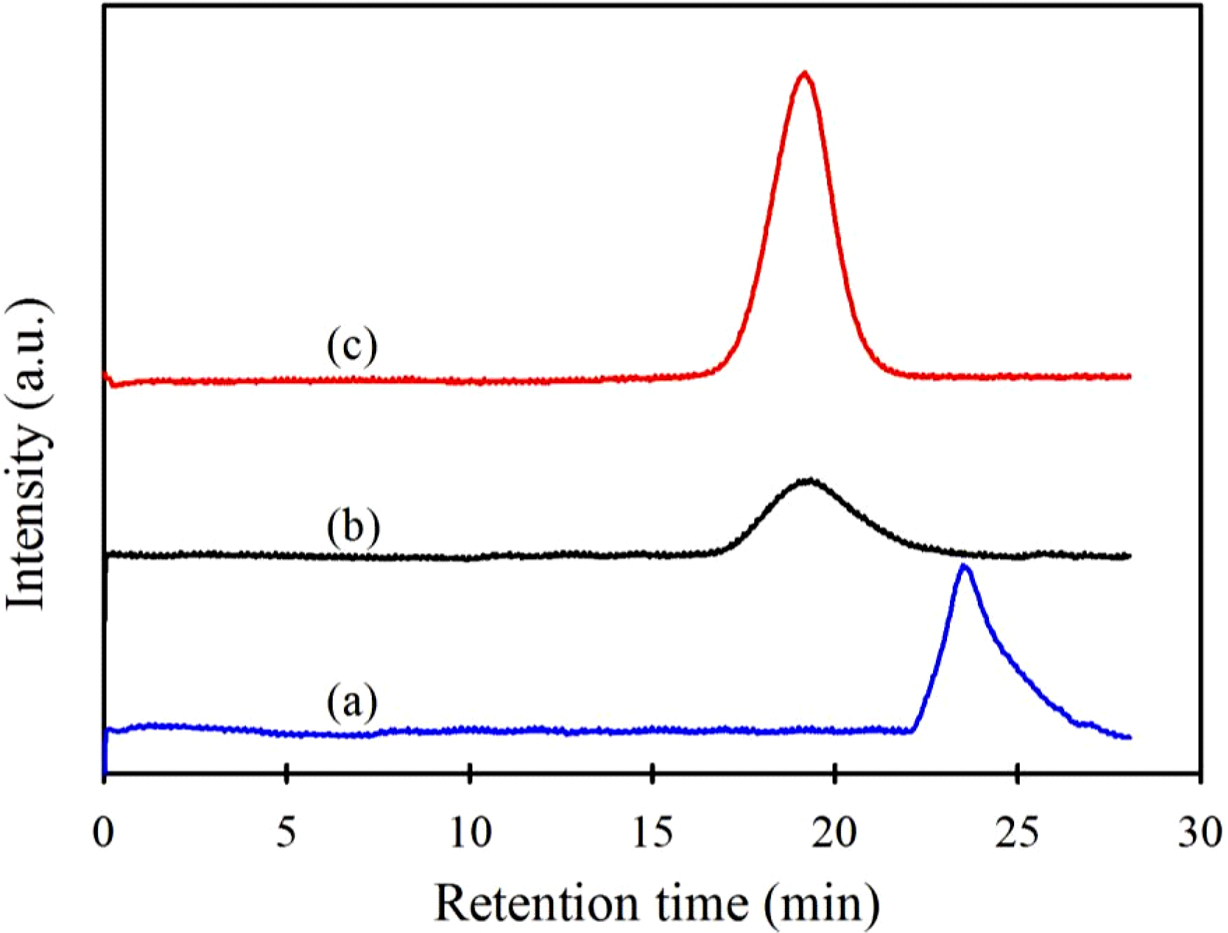
PLA가 제대로 제거되었는지 여부는 GPC 크로마토그램을 얻어 확인하였다. Figure 2는 CA, 본 연구에서 합성한 PLA 및 CA-g(38.4)-PLA의
GPC 크로마토그램을 나타낸 것이다. 이를 보면 출발 물질로 사용한 CA 보다 짧은 머무름 시간에서 피크가 나타나고 있어 CA-g-PLA의
분자량이 CA보다 커진 것을 알 수 있으며, 호모 PLA의 피크가 나타나는 CA보다 머무름 시간이 긴 영역에서 피크가 나타나고 있지 않아 호모
PLA가 포함되어 있지 않음을 알 수 있다.
TGA 분석에 의한 CA-g-PLA의 그래프트율. 그래프트율은 그래프트 공중합체의 특성을 나타낼 때 가장
중요한 요소이다. CA-g-PLA의 그래프트율은 CA 무게에 대해 그래프트된 PLA의 무게 비율로 나타내어지며, 본 연구에서는
TGA 분석에 의한 그래프트율이 xx%인 시료를 CA-g(xx)-PLA로 명명하였다. 이러한 그래프트율은
그래프트시키기 전의 CA 무게와 그래프트시킨 후 얻어진 물질의 무게비로부터 구할 수 있지만, 시료 제조 및 침전, 수세 과정에서 무게 손실이 발생하기
때문에 단순히 반응 전후 무게로부터 그래프트율을 결정하기는 곤란하다.
이러한 경우 얻어진
공중합체를 기기분석하여 그 결과로부터 그래프트율을 구할 수 있다. 본 연구에서는 CA-g-PLA 시료의 TGA 곡선 및 1H NMR 스펙트럼을 분석하여 그래프트율을 구하고 그 값들을
상호 비교하였다. 이같이 TGA 또는 NMR 분석으로부터 그래프트율을 구하는 방법은 여러 문헌에 나타나고 있다.7,21,22
NMR 분석법을 사용하면 그래프트율 뿐만 아니라 CA-g-PLA에서 PLA의 몰치환도(molar substi-tution, MS), 락틸기 치환도(lactyl degree
of substitution, lactyl DS), 그래프트된 PLA의 중합도 등을 함께 구할 수 있다.7,21,22
먼저 CA-g-PLA의
TGA 열분해 곡선을 CA와 PLA 호모 고분자의 열분해 곡선과 비교함으로써 CA-g-PLA의 그래프트율을 구하였다. CA,
PLA, CA-g-PLA의 TGA 열분해 곡선을 Figure 3에 나타내었다. 이를 보면 호모 PLA의 열분해 개시온도는
239 °C에서, CA의 열분해 개시온도는 327 °C에서 나타나고 있으며, CA-g-PLA
시료는 2 단계의 열분해 거동을 나타내고 있다. 이 CA-g-PLA 열분해 곡선으로부터, 상대적으로 열분해 온도가 낮은 PLA 가지사슬이
먼저 분해된 후에 CA 주사슬의 열분해가 순차적으로 일어남을 알 수 있다. 또한 CA-g-PLA에서 CA 주사슬의 열분해 온도가 호모
CA보다는 낮아지고 PLA 가지사슬의 열분해 온도가 호모 PLA보다는 높아지는 것을 통해, CA 주사슬의 열분해가 PLA 가지사슬의 영향을 받으며
동시에 PLA 가지사슬도 CA 주사슬의 영향을 받는다는 것을 알 수 있다. 이러한 결과는 본 연구에서 얻은 CA-g-PLA 시료가
단순히 CA와 호모 PLA의 혼합물이 아니라는 것을 나타내고 있어 CA 주사슬에 PLA가 잘 그래프팅되었음을 간접적으로 알려준다. 뿐만 아니라
CA-g-PLA의 경우 250 °C 이하에서 호모 PLA의 분해에 의한 무게 감소가 나타나고 있지 않아, CA-g-PLA 시료에서
호모 PLA가 잘 제거되어 순수한 CA-g-PLA를 얻었음을 확인할 수 있다.
CA-g-PLA
시료의 열분해 곡선의 변곡점을 기준으로 PLA와 CA 무게비를 구하였다. CA의 열분해는 약 327 °C에서 시작되므로 그 이전의 온도에서 열분해가 되는
것은 PLA 가지사슬이라고 판단할 수 있다. 이를 근거로 하여 CA 무게에 대한 PLA 무게를 백분율로 나타내어 CA-g-PLA 시료의
그래프트율로 하였다. 이때 단순히 변곡점에서의 값을 사용하면 곤란하다. 이는 CA와 PLA 모두 500 °C까지 열분해시킨 후의 잔존량이 0이 아니며, 이때의
잔존 값이 두 고분자가 서로 다르기 때문이다. 일단 변곡점까지의 감소무게는 PLA에 기인한다고 하더라도 변곡점 이후에도 PLA 분해가 포함될 수
있다. 500 °C까지 열분해시킨 CA-g-PLA 시료의
잔존량이 CA의 잔존량보다 더 작은 값을 나타내는 것은 그만큼 PLA가 더 분해되었음을 나타낸다. 또한 500 °C에서 PLA의 잔존량이 0이 아니므로 구해진 감소무게에서
PLA의 최종분해율을 보정해 주어야 한다.
이 같은 방법으로
L-락타이드:CA 무게 비율을 14:1로 하여 130 °C에서 16시간 반응시켜 얻은 Figure 3(c) 시료에 대해서 변곡점에서의 값과 위에서 설명한
보정 방법을 사용하여 그래프트율을 구한 결과 34.4%이었으며, 앞에 설명한 바와 같이 이 시료를 CA-g(34.4)-PLA로
표기하였다.
그래프트 반응 시
CA에 대한 L-락타이드 무게 비율과 반응 시간을 달리하여 여러 CA-g-PLA 시료들을 얻고 이들의 TGA 열분해 곡선으로부터
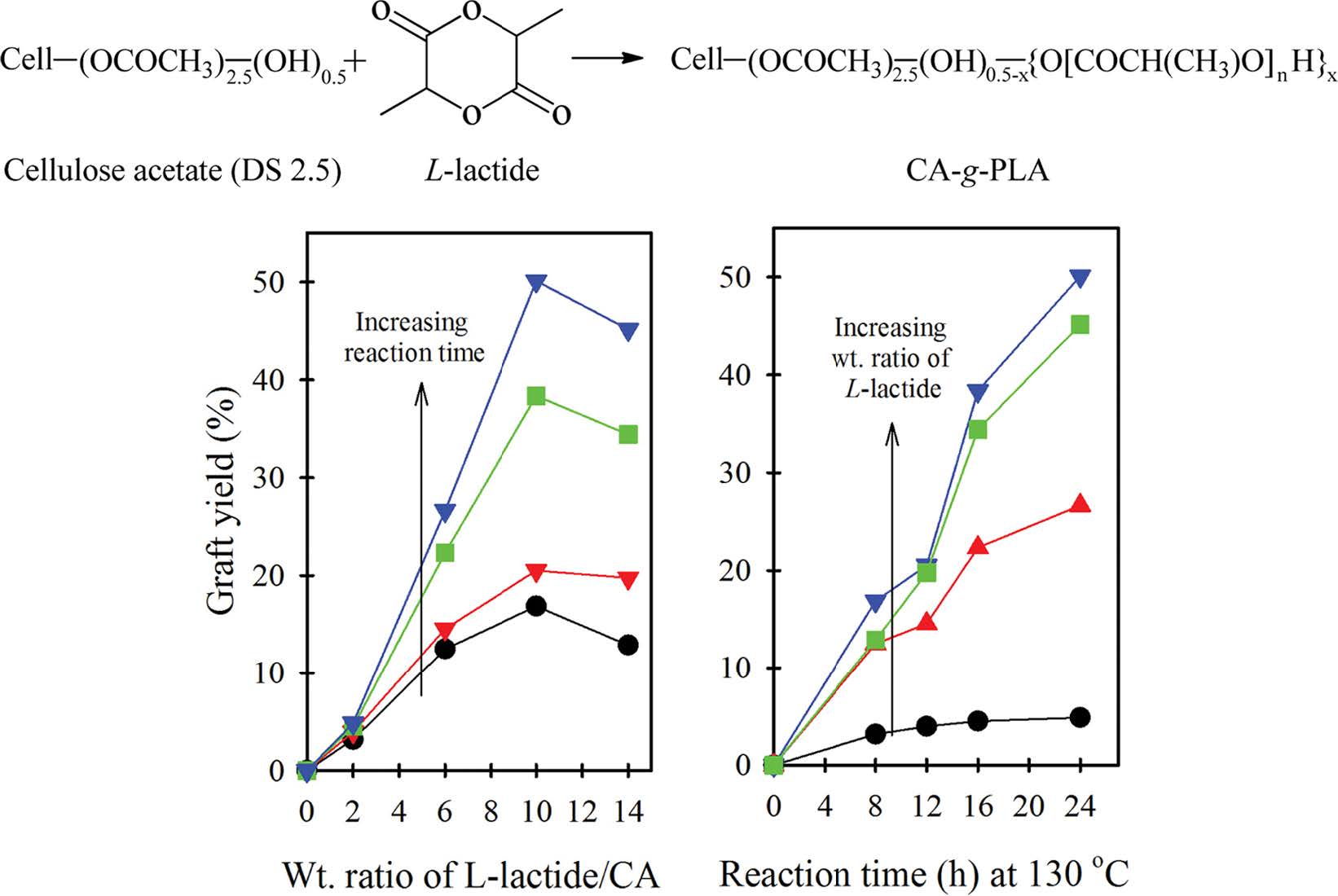
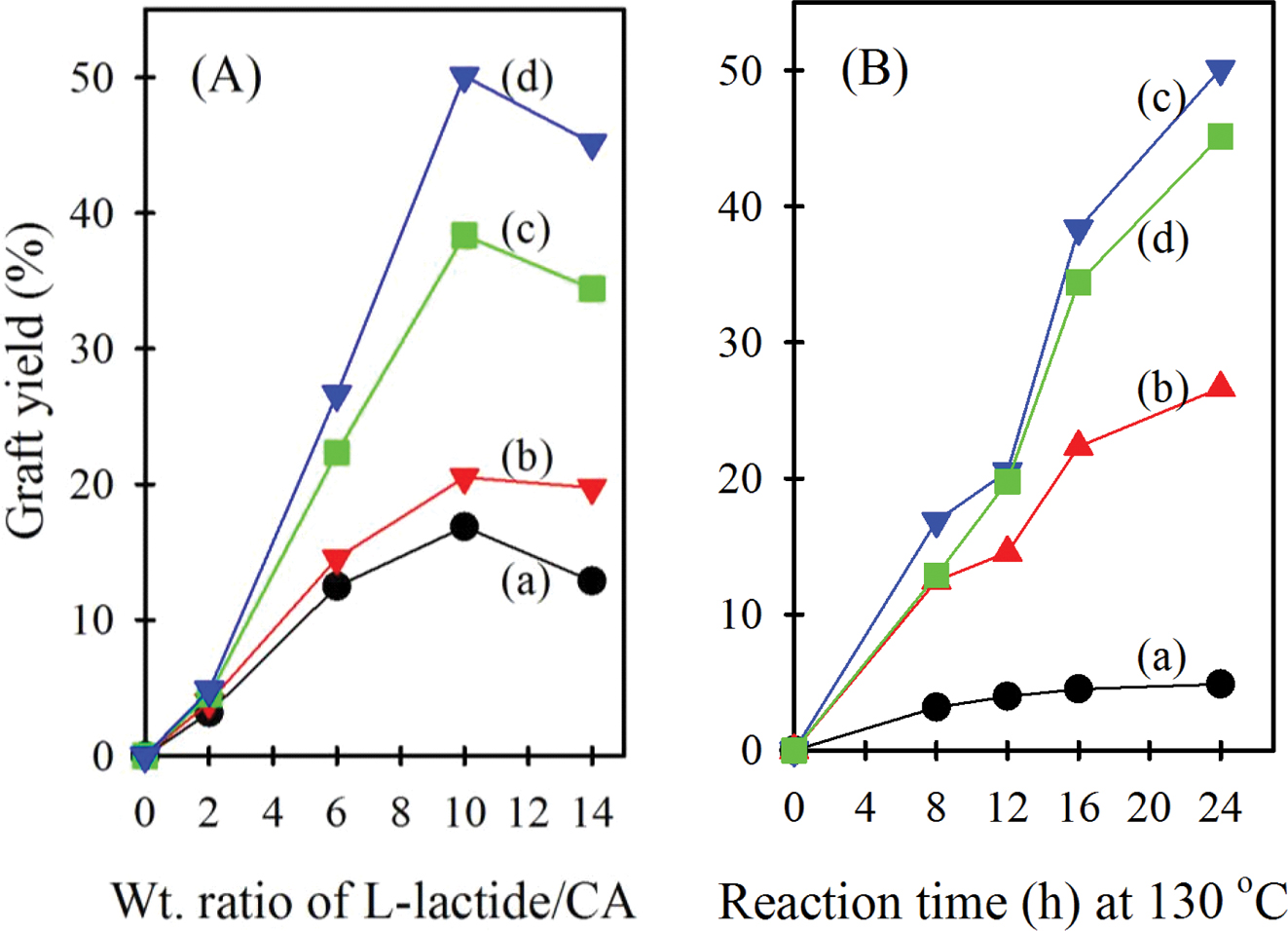
그래프트율을 구한 뒤, CA와 L-락타이드 무게 비와 반응 시간이 그래프트율에 미치는 영향을 Figure 4에 나타내었다.
Figure 4(A)는 130 °C에서 8, 12, 16, 24시간 동안 반응시킨 시료들의 그래프트율을 CA에 대한 L-락타이드의
무게비에 대해서 나타낸 것이다. CA에 대한 L-락타이드의 무게 비율이 커질수록 CA-g-PLA의 그래프트율이 증가하지만
그 비율이 10:1 이상이 되면 그래프트율이 오히려 감소하는 경향을 나타내었다. 본 연구에서는 CA-g-PLA 합성 시 촉매의 양은
L-락타이드의 0.2 wt%로 하여 L-락타이드 무게에 비례해서 넣어주었다. 따라서 L-락타이드의 양이 많아지면
촉매의 양도 많아지지만 그래프팅이 될 수 있는 CA 주사슬의 히드록시기에는 제한이 있다. 이 경우 L-락타이드 양이 어느 정도 이상으로
되면 그래프트되는 것보다 상대적으로 호모 PLA가 많이 생성되어 그래프트율이 오히려 감소하는 경향을 나타낸다고 판단하였다. Stolt,23
Lee,24,25 John26 등에 의하면 L-락타이드의
개환반응에서 고분자 사슬의 성장이 마지막 단계에 다다랐을 때 단량체의 농도가 낮으면 백 바이팅, 사슬 내부의 에스터 교환반응과 사슬간의 사슬 재분배에
의해 해중합이 일어난다. CA에 대한 L-락타이드의 양이 지나치게 많을 경우에는 상대적으로 호모 PLA의 생성이 많아지고 남아 있는
단량체의 농도도 낮아져 해중합이 일어날 가능성도 있다. 뒤의 NMR 결과에서 설명하겠지만, CA-g-PLA에 그래프트된 PLA의 중합도는
매우 작기 때문에 락타이드 양이 어느 정도 이상이 되면 그래프트율이 일정해지는 경향은 그래프트된 PLA의 해중합에 의한 것이기 보다는 CA에 있는
히드록시기에서 시작되는 그래프트점이 한정되어 있어서 L-락타이드 양이 10:1 이상으로 커지면 그래프트율이 일정해지는 것으로 판단된다.
이 같이 L-락타이드 양이 많아질 때 그래프트율이 더 이상 증가하지 않고 감소하는 현상은 단량체와 촉매의 비를 조절함으로 개선이 가능함을
추가 실험을 통해 확인한 바 있다. 촉매의 비율을 크게 하면 그래프트율이 더 커지며, 실험 범위 내에서 락타이드 양이 많아지더라도 그래프트율이
감소하지 않고 계속 증가하는 경향을 나타내었다.
Figure 4(B)는
CA에 대한 락타이드 비율이 일정할 때 TGA 분석에서 구한 그래프트율 변화를 130 °C에서 반응시키는 시간에 대해서 나타낸 것이다. 이를
보면 반응 시간이 길어질수록 CA-g-PLA의 그래프트율이 점차 증가한다. 또한 CA에 대한 L-락타이드의 무게비가
10:1일 때와 14:1일 때의 값이 큰 차이를 보이지 않음도 확인할 수 있다. 그러나 반응 시간을 24시간으로 할 경우 얻어진 시료가 상당히
심하게 변색되었을 뿐만 아니라 반응 후 내용물의 점도가 너무 커져서 이를 침전시키기 위해서는 추가적인 용매를 사용하거나 분쇄를 해주어야 하는 등의
추가적인 공정이 필요하였다. 이는 공업적인 응용에 적합하지 않기 때문에 반응 시간은 16시간이 적합하다고 판단하였다.
이러한 결과들로부터
CA-g-PLA 합성에 가장 효율적인 합성 조건은 CA에 대한 L-락타이드 무게비를 10:1로 하면서 130 °C에서 16시간 동안 반응시키는 것이라고 판단하였다.
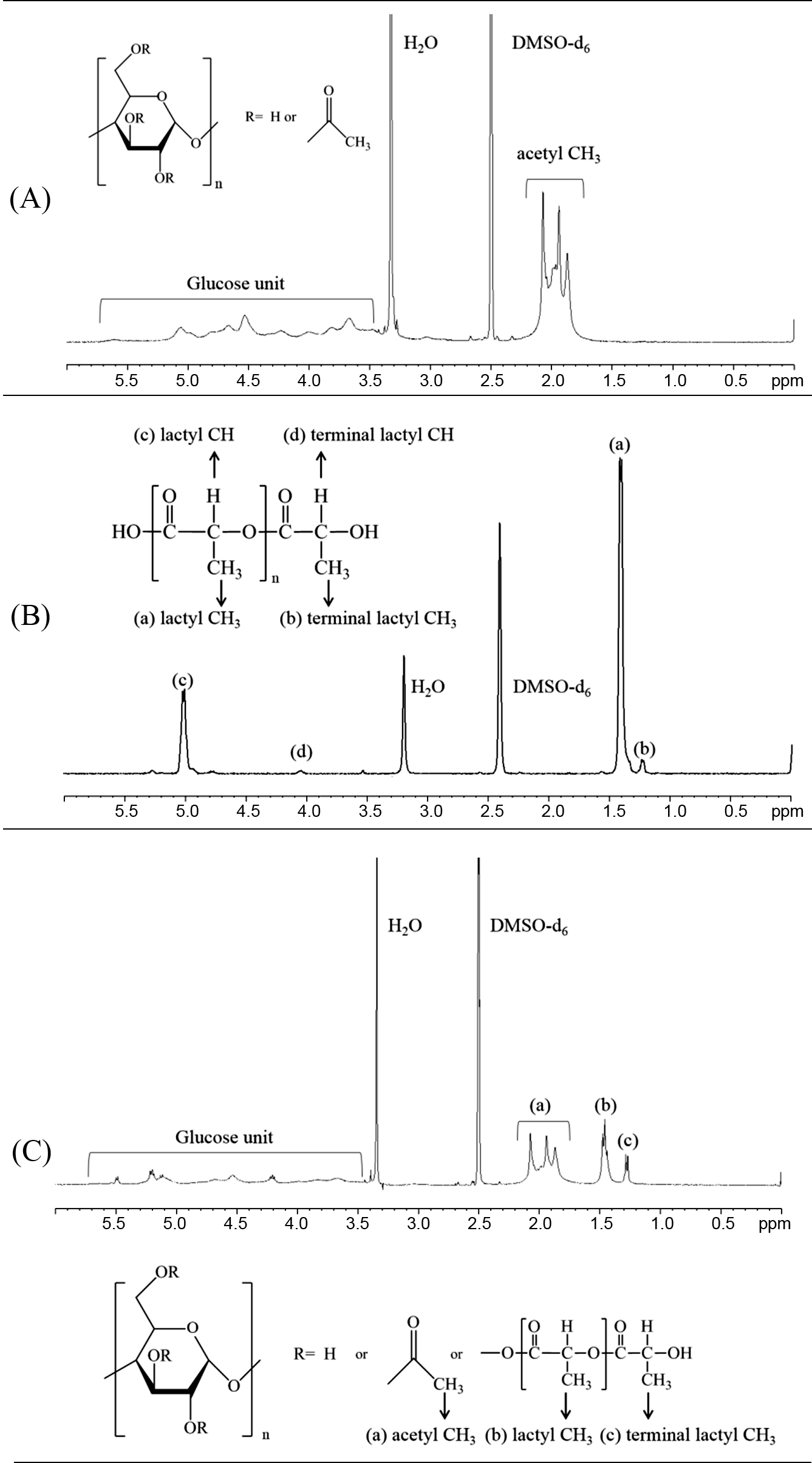
1H NMR을 사용한 CA-g-PLA의 그래프트율 분석. Figure 5(A), (B), (C)는 각각 CA, 호모 PLA, CA-g(38.4)-PLA의 1H
NMR 스펙트럼을 나타낸 것이다. CA의 스펙트럼에서 2.072, 1.940, 1.871 ppm에서 나타나는 피크는 CA 아세틸기의 -CH3에
기인하는 것이고, 5.631, 5.059, 4.532, 3.670 ppm은 글루코스 유닛에 기인한다.27,28 호모
PLA의 경우 1.478, 1.455, 1.441, 1.437 ppm은 락틸기의 -CH3, 1.285, 1.270 ppm은 락틸기 말단의 -CH3에 해당하며, 기타
5.212, 5.196 ppm에서 락틸기 -CH에, 4.197 ppm에서 락틸기 말단의 -CH에 기인한 피크가 나타나고
있다.29,30 한편, CA-g-PLA의 스펙트럼에서는
2.072, 1.939, 1.868 ppm에서 CA 아세틸기의 -CH3, 1.474, 1.456, 1.443, 1.438 ppm에서 락틸기의 -CH3,
1.284, 1.267 ppm에서 락틸기 말단의 -CH3가 각각 나타나며 글루코스 유닛에 기인한 피크는 5.500,
5.199, 4.676, 3.667 ppm에서 나타나고 있다.21,22,27-30
Figure 5(C) 스펙트럼에서
CA 아세틸기의 -CH3(2.072, 1.939, 1.868 ppm)와 PLA 가지사슬 내부에 있는 락틸기 -CH3
(1.474, 1.456, 1.443, 1.438 ppm), PLA 가지사슬 말단에 있는 락틸 -CH3 (1.284, 1.267 ppm)의 적분값을 구하여 식 (1)-(3)에 대입하면
그래프트된 PLA의 몰 치환도, 락틸기 치환도, 그래프트된 PLA 가지사슬의 평균 중합도를 구할 수 있다.21,22 또한 그래프트된
PLA의 몰 치환도를 알면 CA 반복 단위의 분자량, 락틸기 유닛의 분자량으로부터 그래프트율을 구할 수 있다.21,22 식
(4)에서 267(g/mol)은 CA의 반복단위 분자량을, 72 (g/mol)은 락틸기 유닛의 분자량을 의미한다.
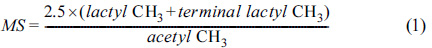


위 식에서 몰 치환도와
락틸기 치환도는 글루코스 유닛 하나를 기준으로 구한 후, 알고 있는 값인 아세틸 치환도 2.5를 곱해주어 전체 몰 치환도와 락틸기 치환도를 구한
것이다. 몰 치환도는 CA 1 몰 당 락틸기가 몇 몰 치환되었는 지를 나타내는 값이다. 락틸기의 -CH3와 락틸기 말단의
-CH3의 적분비 값을 더한 후 아세틸기의 -CH3의 적분비로 나누어주면 AGU 당 락틸기가 몇 몰 치환되었는지
구한 후, CA의 아세틸 치환도인 2.5를 곱해주어 구할 수 있다. 락틸 치환도는 셀룰로오스 반응기 중 몇 개가 락틸기로 치환되었는지를 나타내는
값이기 때문에, 몰 치환도와 달리 락틸기 -CH3는 제외하고 락틸기 말단의 -CH3의 적분비 만을 이용하여
구한다. 그래프트된 PLA 가지사슬의 중합도는 PLA 가지사슬 하나에 몇 개의 락틸기가 중합되었는지를 의미하며, 몰 치환도를 락틸 치환도로 나누어
주면 PLA 가지사슬의 평균 중합도를 구할 수 있다. CA-g-PLA의 그래프트율을 구할 때 이용되는 CA-g-PLA의
PLA 무게함량은 CA 반복단위 분자량인 267 g/mol과 락틸기 유닛의 분자량인 72 g/mol을 이용하여 합성된 CA-g-PLA의
분자량과 그래프트된 PLA 가지사슬의 분자량으로 구할 수 있다.21,22
이러한 방법에 의해
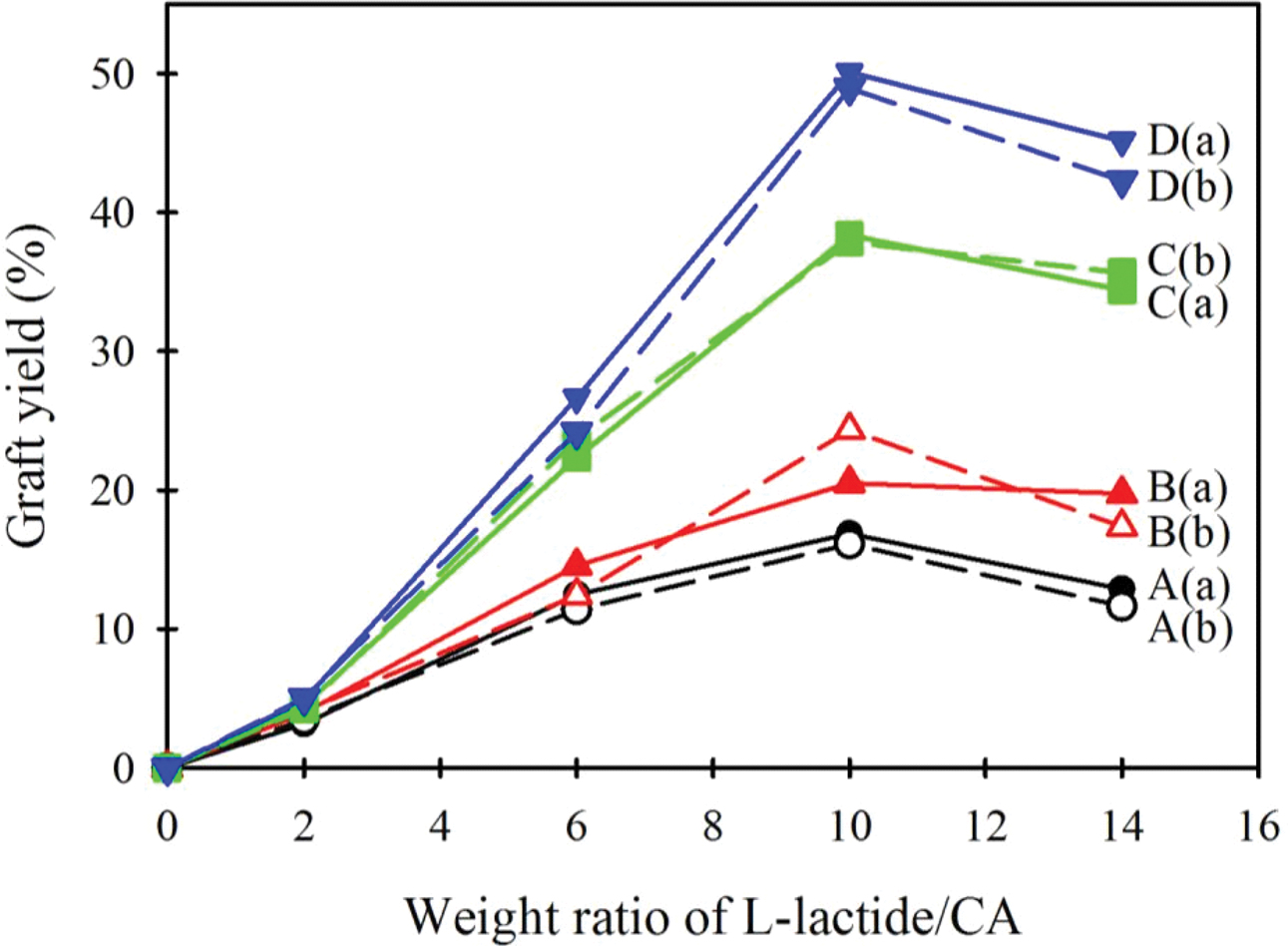
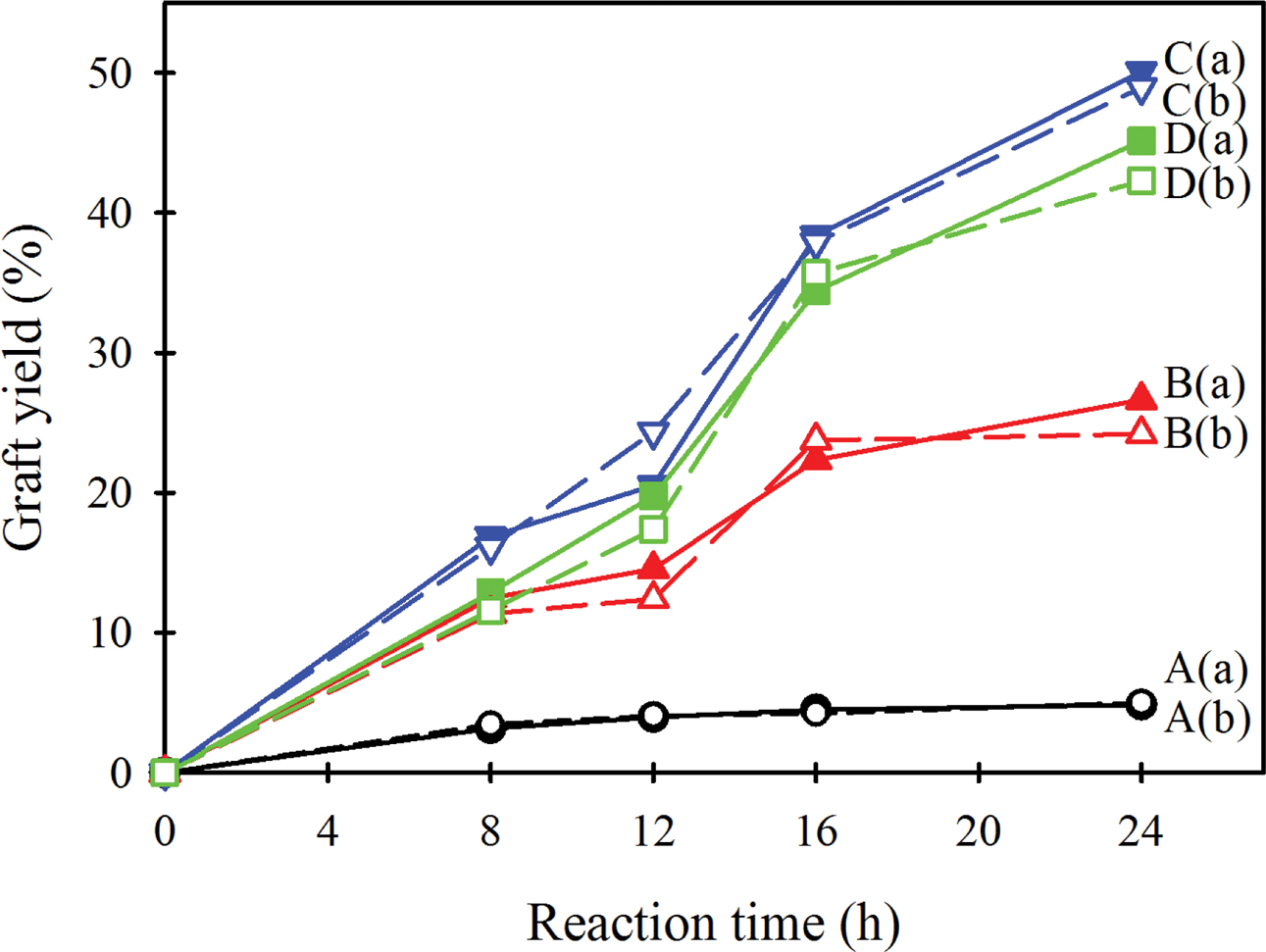
NMR 분석으로부터 구한 각 CA-g-PLA의 그래프트율을 TGA로부터 구한 그래프트율과 함께 Figure 6과
Figure 7에 나타내었다. 이들 결과를 보면 전혀 다른 두 가지 분석 방법인 TGA와 NMR 데이터에서 구한 그래프트율이
거의 일치하고 있으며, 이는 두 가지 실험 결과 모두에 대한 신뢰성을 부여하는 것이라고 할 수 있다.
Figures 6과 7을
보면 TGA 분석을 통해 얻은 결과와 마찬가지로, CA에 대한 L-락타이드의 무게비가 10:1 이하일 때는 무게비가 증가함에 따라
그래프트율이 증가하지만 그 이상에서는 그래프트율이 거의 일정하거나 약간 감소하며, 반응 시간이 길어질수록 그래프트율이 증가한다는 것을 확인할 수
있다.
한편, NMR 스펙트럼
결과를 (1)-(4) 식에 대입하여 구한 CA에 대한 L-락타이드 무게비에 따른 CA-g-PLA의 몰치환도, 락틸 치환도,
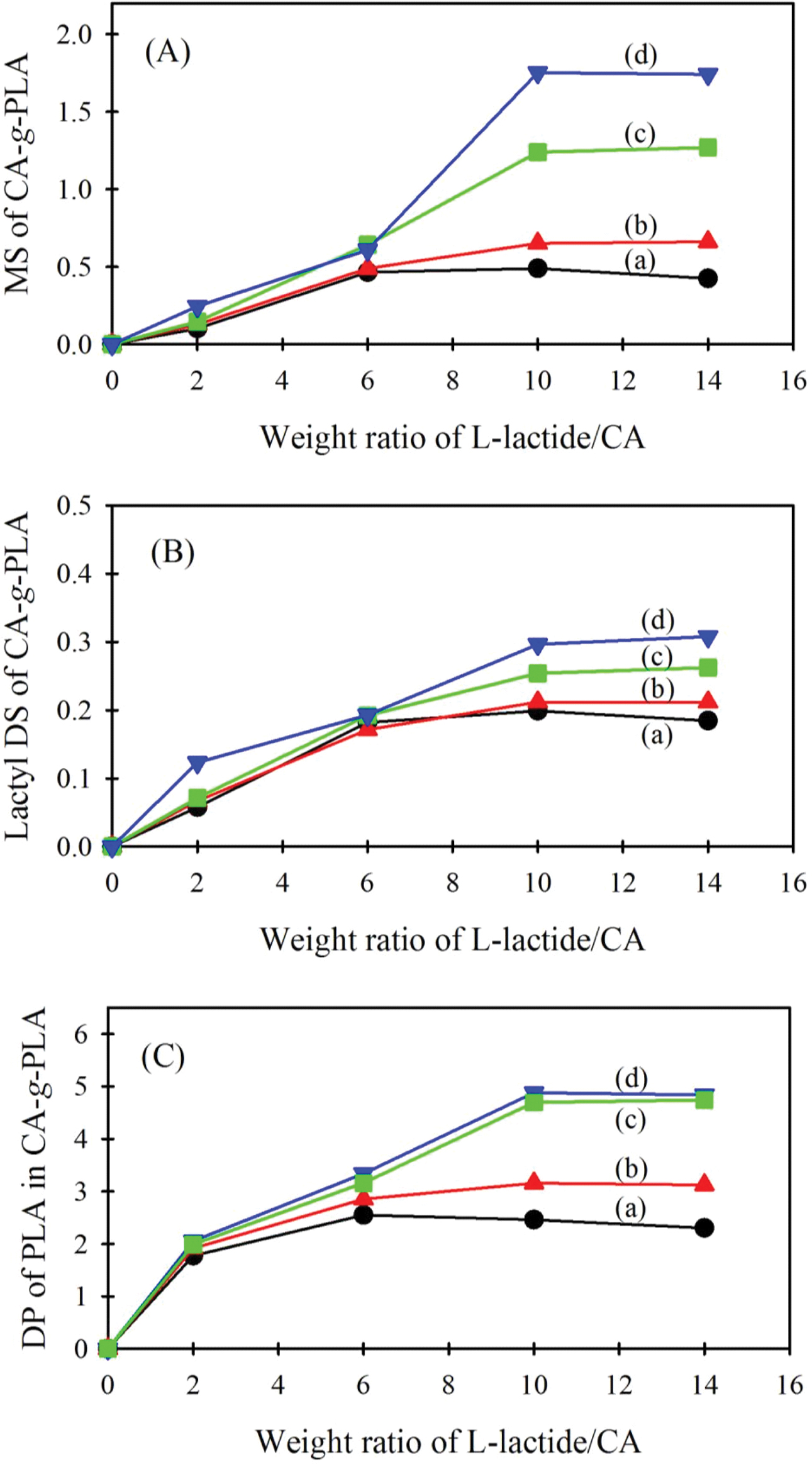
그래프트된 PLA 가지사슬의 중합도 변화를 각각 Figure 8(A), (B), (C)에 나타내었다. 이들 결과를 보면 CA에 대한 L-락타이드
비율이 10:1 이상으로 되면 PLA의 몰 치환도가 거의 일정하게 되고, 락틸 치환도는 소폭 증가하여, 그래프트된 PLA 중합도가 일정해지는 경향을 나타낸다. CA-g-PLA에 CA 1몰 당 그래프트된
락틸기의 몰 수가 커지면 CA 주사슬에 그래프팅 사이트가 많아지고, PLA 가지사슬에 중합된 락틸기의 개수도 커지면서 그래프트율이 증가된다. L-락타이드의
개환반응은 매우 빠른 반응으로 초기에 많은 호모 PLA가 중합된다. 촉매의 양은 많아졌지만 CA의 양은 변하지 않아 락틸 치환도가 조금 증가하기는
하지만 그래프트가 일어날 수 있는 반응기 수에는 한계가 있어 호모 PLA의 중합이 보다 많이 일어난다. 따라서 L-락타이드 양이 어느
정도 이상으로 커지면 여러 값들이 증가하지 않는 결과는 L-락타이드 양에 비례하여 많아진 촉매의 영향으로 판단된다.
반응 시간에 따른
CA-g-PLA의 몰 치환도, 락틸 치환도, 그래프트된 PLA 가지사슬의 중합도 변화를 각각 Figure 9(A), (B), (C)에 나타내었다. 반응 시간 16시간 이후부터는 몰
치환도와 락틸 치환도 값은 증가하였으나 PLA 가지사슬 중합도에는 큰 차이가 없었다. 이를 통해 반응 시간이 길어질수록 새로운 활성점이 생겨
PLA가 그래프트되며 이에 따라 몰 치환도는 증가하지만, 반응 시간 16시간 이상에서는 가지사슬에 그래프트된 락틸기 수에는 큰 변화가 없는 것을 확인하였다. 이 때 주목할 만한 점은 약제 비율이 10:1 인 경우의
값들과 약제 비율이 14:1일 때의 값들이 큰 차이를 보이지 않으며 서로 비슷한 값들을 나타낸다는 것이다. 이는 약제 비율이 10:1 이상부터는
L-락타이드 증가에 따른 영향이 크지 않다는 것을 재확인해주고 있다.
이상의 실험 결과들은
CA-g-PLA를 합성할 때 변화시킨 실험 조건(락타이드 양, 반응 시간)에 따른 여러 가지 변화를 나타낸 것이지만, 치환도나 중합도는
반응 조건 자체보다는 그래프트율에 의존된다. 이에 따라 각 CA-g-PLA 시료들의 그래프트율(TGA 분석에 의해 구한 값)에 따른
몰 치환도, 락틸기
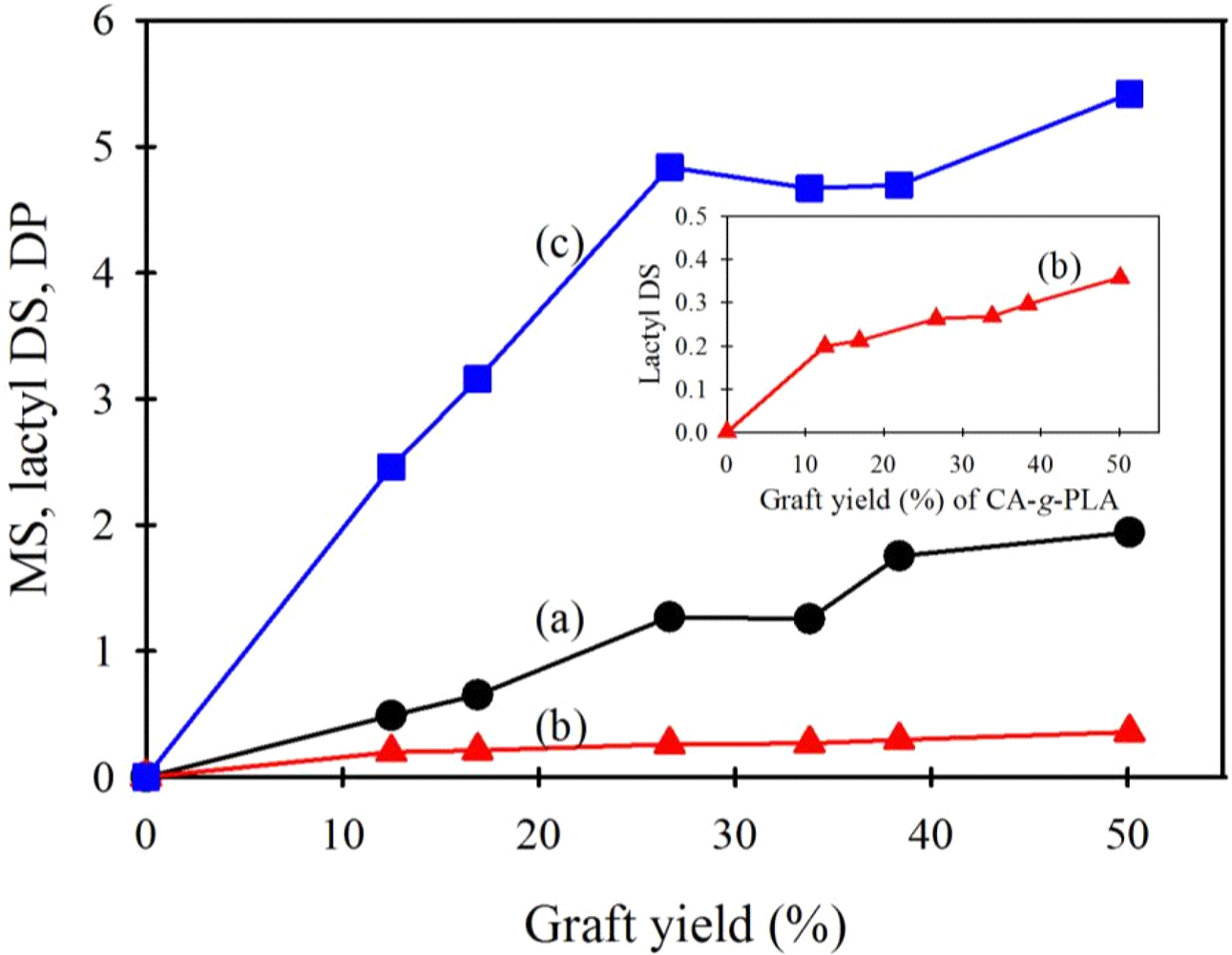
치환도, 그래프트된 PLA 가지사슬 중합도 변화를 Figure 10에 나타내었다. 이를 보면 그래프트율이 증가할수록
세 값이 모두 증가하고 있다. 이때, 몰 치환도와 락틸기 치환도는 그래프트율이 커지면 거의 직선적으로 증가하고 있지만, 그래프트된 PLA의 중합도는
그래프트율 27% 이상에서 그 값이 거의 비슷하거나 증가 정도가 작아졌다. 이는 그래프트율이 어느 정도 커지는 이유가 몰 치환도 및 락틸 치환도가
커지면서 그래프트된 가지의 길이도 증가되는 것 때문이지만, 어느 정도 그래프트율이 커지면 그래프트된 PLA 가지의 길이는 거의 비슷하면서 몰 치환도 또는 락틸 치환도가 커져 그래프트율이 커진다는
것을 나타낸다.
한편, Figure 10(b)를
보면 락틸기 치환도는 최대 약 0.32 의 값을 갖는다. 합성에 사용된 CA의 아세틸기 치환도는 2.5 이기 때문에 히드록시기 치환도는 AGU
당 0.5이다. PLA 가지사슬은 CA 주사슬의 히드록시기에 그래프트되기 때문에 락틸기 치환도가 0.32라는 것은 히드록시기 0.5 몰 중에서
0.32 몰이 그래프트되어 CA 주사슬의 최대 64%의 히드록시기에 PLA가 그래프팅되었음을 알 수 있으며, 이에 따라 Figure 1(d)의
IR 스펙트럼에서 히드록시기 밴드 흡광도 크기가 크게 감소하였음을 알 수 있다.
이상의 결과들로부터
그래프트시키는 시간을 길게 할수록 치환도 등이 커지고, CA에 대한 L-락타이드의 무게비는 10:1 이상으로 되면 L-락타이드의
양에 대한 영향을 거의 받지 않는다는 것을 다시 확인할 수 있다.

|
Figure 1 FTIR spectra of (a) L-lactide; (b) PLA; (c) cellulose acetate (CA); (d) CA-g(50.1)-PLA. |
|
Figure 2 GPC chromatograms of (a) PLA; (b) CA; (c) CA-g(38.4)-PLA. |

|
Figure 3 TGA curves of (a) CA; (b) PLA; (c) CA-g(34.4)-PLA. |
|
Figure 4 Changes in the graft yields of CA-g-PLA obtained by TGA analysis (A) with increasing weight ratio of L-lactide to CA; (B) with increasing reaction time at 130 °C. (A) Reaction time at 130 °C: (a) 8 h; (b) 12 h; (c) 16 h; (d) 24 h; (B) weight ratio of L-lactide to CA: (a) 2:1; (b) 6:1; (c) 10:1; (d) 14:1. |
|
Figure 5 1H NMR spectra of (A) CA; (B) PLA; (C) CA-g(38.4)-PLA. |
|
Figure 6 Comparison of the graft yields obtained by (a) TGA analysis; (b) 1H NMR analysis. Reaction time at 130 °C: (A) 8 h; (B) 12 h; (C) 16 h; (D) 24 h. |
|
Figure 7 Comparison of the graft yields obtained by (a) TGA analysis; (b) 1H NMR analysis. Weight ratio of L-lactide to CA: (A) 2:1; (B) 6:1; (C) 10:1; (D) 14:1. |
|
Figure 8 Changes in (A) mol substitution (MS); (B) lactyl DS; (C) DP of PLA in CA-g-PLA with increasing weight ratio of L-lactide to CA. Reaction time at 130 °C: (a) 8 h; (b) 12 h; (c) 16 h; (d) 24 h. |

|
Figure 9 Changes in (A) mol substitution (MS); (B) lactyl DS; (C) DP of PLA in CA-g-PLA with increasing reaction time at 130 °C. Weight ratio of L-lactide to CA: (a) 2:1; (b) 6:1; (c) 10:1; (d) 14:1. |
|
Figure 10 Increases in (a) mol substitution (MS); (b) lactyl DS; (b) DP of PLA in CA-g-PLA with increasing graft yield. |
혼화성이 없는
CA와 PLA 고분자의 상용화제로 사용하기 위하여 여러 가지 그래프트율을 갖는 CA-g-PLA를 합성한 후 이들의 그래프트율과 여러
가지 특성들을 분석하여 다음과 같은 결론들을 얻었다.
L-락타이드의 개환중합을 통해 CA-g-PLA
공중합체를 합성할 수 있으며, 그래프팅 시 CA와 L-락타이드 비율 및 반응 시간을 달리함으로써 그래프트율이 다른 CA-g-PLA를
얻을 수 있다. 본 연구 결과 범위 내에서, 그래프팅시키는 온도 130 °C, CA에 대한 L-락타이드의 비율
10:1, 반응 시간 16 시간의 반응 조건이 가장 효율적이다. CA-g-PLA의 TGA 열분해 곡선과 1H NMR 스펙트럼을 분석하여 그래프트율을 구할 수 있으며,
두 분석에 의한 그래프트율 결과와 거의 일치한다. 촉매를 L-락타이드에 대해 일정 비율(0.2 wt%)을 사용하는 경우 그래프팅 시 사용하는 L-락타이드
양이 많아지면 그래프트율이 증가하지만, 그 비율이 어느 정도 이상으로 되면(본 연구의 경우 10:1) 그래프트율이 더 이상 증가하지 않는다.
NMR 분석에 의하여 몰 치환도, 락틸기 치환도, 그래프트된 PLA 가지사슬의 중합도를 구할 수 있으며, L-락타이드의 양과 반응
시간에 따른 이들 값의 증가는 그래프트율 증가와 일치한다. 그래프트율이 어느 이상으로 되면 그래프트되는 PLA 가지사슬의 중합도 증가가 아니라
그래프트되는 치환점이 많아져 그래프트율이 커진다.
- 1. A. K. Mohanty, A. Wibowo, M. Misra, and L. T. Drzal, Polym.Eng. Sci., 43, 1151 (2003).
-
- 2. C. R. Albarez-Chabez, S. Edwards, R. Moure-Eraso, and K. Geiser, J. Clean.Prod., 23, 47 (2012).
-
- 3. V.P. Ghiya, V. Dave, R. A. Gross, and S. P. Mccarthy, J. Macromol. Sci.,33, 627 (2006).
-
- 4. H. M. Park, M. Misra, L. T. Drzal, and A.K. Mohanty, Biomacromol., 5, 2281 (2004).
-
- 5. K. M. Nampoothiri, N. R. Nair, and R. P. John, Biores.Tech., 101, 8493 (2010).
-
- 6. A.Kowalski, A. Duda, and S. Penczek, Macromolecules, 33, 7359(2000).
-
- 7. N.Ogata, T. Tatsushima, K. Nkane, K. Sasaki, and T. Ogihara, J. Appl. Polym.Sci., 85, 1219 (2002).
-
- 8. R.Quintana, O. Persenaire, Y. Lemmouchi, L. Bonnaud, and P. Dubois, Eur.Polym. J., 57, 30 (2014).
-
- 9. L.Nikolic, I. Ristic, B. Adnadjevic, V. Nikolic, J. Jovanovic, and M. Stankovic, Sensors,10, 5063 (2010).
-
- 10. H.R. Kricheldorf, I. Kreiser-Saunders, and C. Boettcher, Polym., 36,1253 (1995).
-
- 11. S.H. Kim, Y. K. Han, Y. H. Kim, and S. I. Hong, Macromol. Chem. Phys., 193,1623 (1992).
-
- 12. D. Garlotta, J. Polym. Environ., 9, 63(2001).
-
- 13. H.Qian, A. R. Wohl, J. T. Crow, C. W. Macosko, and T. R. Hoye, Macromolecules,44, 7132 (2011).
-
- 14. F. Khabbaz, S. Karlsson, and A. Albertsson, J.Appl. Polym. Sci., 78, 2369 (2000).
-
- 15. O.Dechy-Cabaret, B. Martin-Vaca, and D. Bourissou, Chem. Rev., 104,6147 (2004).
- 16. S.Monga, A. Kaushik, and B. Gupta, Polym. Plast. Tech. Eng., 55,1819 (2016).
-
- 17. C.D. C. Erbetta, R. J. Alves, and R. F. Freitas, J. Biomater. Nanobiotech., 3,208 (2012).
-
- 18. V. Katiyar and H. Nanavati, Polym. Chem., 1,1491 (2010).
-
- 19. D.Zavastin, I. Cretescu, M. Bezdadea, and J. Savic, Colloid Surf. A:Physicochem. Eng. Aspects, 370, 120 (2010).
-
- 20. S.R. Dods, O. Hardick, B. Stevens, and D. G. Bracewell, J. Chromatogr. A, 1376,74 (2015).
-
- 21. J.Bao, L. Han, G. Shan, Y. Bao, and P. Pan, J. Phys. Chem., 119,12689 (2015).
-
- 22. H. Lonnberg, Q.Zhou, H. Brumer III, and A. Hult, Biomacromol., 7, 2178 (2006).
-
- 23. A. Sodergard and M. Stolt, Poly(lactic acid):Synthesis, Structures, Properties, Processing, and Application, Wiley,N.Y., p 27 (2010).
-
- 24. C.Ho, C. Wang, C. Lin, and Y. Lee, Polym., 49, 3902 (2008).
-
- 25. D.Yoo, D, Kim, and D. Lee, Macromol. Res., 14, 510 (2006).
-
- 26. K.Nampoothiri and R. John, Biores. Tech., 101, 8493 (2010).
-
- 27. K.Kowsaka, K. Okaima, and K. Kamide, Polym. J., 18, 843 (2006).
-
- 28. V.W. Goodlett and H. W. Patton, Polym. Chem., 9, 155 (1971).
-
- 29. S.Jacobsen, P. Degee, P. Dubois, and R. Jerome, Polym. Eng. Sci., 7,1311 (1999).
-
- 30. K.Choi, M. Choi, T. Park, and C. Ha, Eur. Polym. J., 49, 2356(2013).
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2019; 43(6): 852-861
Published online Nov 25, 2019
- 10.7317/pk.2019.43.6.852
- Received on Jun 18, 2019
- Revised on Jul 25, 2019
- Accepted on Sep 10, 2019
Services
Shared
Correspondence to
- Young Ho Kim
-
Department of Organic Materials and Fiber Engineering, Soongsil University, Seoul 06978, Korea
- E-mail: ssyhkim@ssu.ac.kr
- ORCID:
0000-0001-8889-3285
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr