





- Preparation and Properties of Eco-friendly Polyurethane Nanocomposites Using Cellulose Nanocrystals with Amino Group as Fillers
Youna Lee, Seosuk Park, and KiRyong Ha†

Department of Chemical Engineering, Keimyung University, Daegu 42601, Korea
- 아미노기가 도입된 셀룰로오스 나노 결정을 충전제로 사용한 친환경 폴리우레탄 나노복합체 제조 및 물성에 대한 연구
이유나 · 박서숙 · 하기룡†
계명대학교 공과대학 화학공학과
In this study, a polyol
mixture of bio-polyol and petroleum-polyol in a mass ratio of 3:7 was reacted
with 4,4'-methylenebis(phenyl isocyanate) (MDI) to prepare an eco-friendly
polyurethane (PU) nanocomposite. Also, using 3-aminopropyltriethoxysilane (APS)
silane coupling agent, -NH2 group was grafted onto the cellulose
nanocrystals (CNCs) surfaces, which was used as fillers. We studied the effects
of surface modified CNC fillers on the mechanical properties of PU
nanocomposites using thermogravimetric analysis (TGA), universal testing
machine (UTM) and dynamic mechanical analyzer (DMA). The -NH2 groups
grafted on the CNC surface can form urea bonds with -NCO groups of MDI at the
interface, which increased the tensile strength and modulus of PU
nanocomposites more than those of the pristine CNC as fillers.
본 연구에서는 바이오 폴리올과 석유계 폴리올을 질량비 3:7 비율로 혼합한 폴리올을 4,4'-methylenebis (phenyl
isocyanate)(MDI)와 반응시켜 친환경 폴리우레탄 나노복합체를 제조하는 연구를 수행하였다.
또한 3-amino-propyltriethoxysilane(APS) 실란 커플링제를 사용하여, 셀룰로오스 나노 결정(cellulose nanocrystal, CNC) 표면에 -NH2기를 도입하여 충전제로 사용하였다. 표면 개질된 CNC 충전제가 폴리우레탄 나노복합체의 물성에 미치는 영향을 열 중량 분석기(TGA),
만능재료시험기(UTM) 및 동적기계분석기(DMA)를
사용하여 체계적으로 분석하였다. CNC 표면에 도입된 -NH2기는 MDI의 -NCO기와 계면에서 우레아 결합을 형성하여 순수 CNC를 충전제로 사용한 폴리우레탄 나노복합체보다 인장강도와 탄성률이 더 많이 증가되는 것을 확인하였다.
Polyurethane (PU) nanocomposites were prepared by
reacting 4,4'-methylenebis(phenyl isocyanate) (MDI) with
polyol mixed with 30% soybean oil based bio-polyol and 70%
poly(tetrahydrofuran) (PTHF). PU nanocomposites with
3-aminopropyltriethoxysilane (APS) treated cellulose nanocrystal (CNC-APS) as
fillers showed higher Tg than that of cellulose nanocrystal
(CNC) fillers due to strong urea bonding formation between CNC-APS and PU
interface.
Keywords: cellulose nanocrystal (CNC), silane coupling agent, 3-aminopropyltriethoxysilane (APS), polyurethane, bio-polyol
폴리우레탄(polyurethane,
PU)은 폴리올(polyol)과 이소시아네이트(isocyanate)가 반응하여 화학구조 상 우레탄기(-NHCOO-)를 가지는 고분자 화합물이다.1 폴리우레탄은
뛰어난 물성을 가지고 있어서 자동차, 전자 제품, 의류, 가구, 단열재 및 접착제 등 여러 분야에서 널리 사용되고 있다.2 폴리우레탄과
같은 석유화학물질의 사용량은 점점 증가하지만 폐기물 처리로 인해 생기는 환경오염과 한정된 자원 고갈은 심각한 문제로 대두되고 있다. 이러한 문제를
해결하려는 노력들로 식물성 천연유지를 원료로 사용하거나 전분, 리그닌, 셀룰로오스와 같은 바이오 충전제의 활용에 대한 연구가 최근 활발히 진행되고
있다.3-5
식물성 천연유지는
대두유, 피마자유 및 팜유 등을 주로 사용하며, 글리세롤 한 분자에 세 개의 지방산이 결합된 트리글리세라이드(triglyceride) 구조로 이루어져
있다. 대부분의 식물성 기반의 바이오 폴리올(bio-polyol)은 에폭시화/개환 반응을 통하여 -OH기를 가지는 단량체로 전환된다.6
셀룰로오스는 자원이
풍부하고 경제성, 재생가능성 및 친환경성을 가지는 물질로 광범위한 분야에 사용되고 있다. 그 중 셀룰로오스 나노 결정(cellulose
nanocrystal, CNC)은 고강도, 고탄성률과 같은 우수한 기계적 물성과 높은 표면적을 가지고 있어서 나노복합체의 보강 충전제로 사용하기
위한 연구가 활발히 진행 중이다.7,8
CNC는 표면에 다량의
-OH기를 가지는 친수성 물질이기 때문에 소수성의 고분자 물질과 혼합하여 사용하기 위해서는 실란 커플링제를 사용하여 CNC 표면을 개질하여
상용성과 계면 결합력을 높여주어야 한다.9
실란 커플링제는 동일한
분자 내에 2개 이상의 반응기를 가지고 있으며, 그 중 하나는 친수성 무기물과 반응하고, 다른 한 쪽은 소수성 고분자와 반응하여 무기물 충전제와
유기질 고분자간의 다리(bridge) 역할을 하는 물질이다.10 실란 커플링제는 산성이나 알칼리성에서 가수분해가 잘 일어나며,
산성에서는 가수 분해된 단량체 형태가 안정하고, 알칼리성에서는 축합 반응이 일어난 올리고머(oligomer) 형태가 안정한 것으로 알려져 있다.11
본 실험에서는 아미노기(amino
group, -NH2)를 가지고 있는 실란커플링제인 3-aminopropyltriethoxysilane(APS)를 사용하여
CNC를 개질하여 CNC-APS를 합성하고자 한다. PU 나노복합체 제조 과정에서 CNC 표면에 도입된 -NH2기와 4,4'-methylenebis(phenyl
isocyanate)(MDI)의 -NCO기가 계면에서 우레아 결합을 생성할 수 있다. 우레아 수소 결합의 길이(4.63 Å)가 우레탄 수소 결합의 길이(5.10 Å)보다 짧기 때문에 우레아 수소 결합의 강도는 우레탄
수소 결합의 강도보다 높은 것으로 알려져 있다.12 따라서 우레아 결합 사이의 수소결합이 우레탄 결합 사이의 수소 결합보다
강하기 때문에 더 강한 경질 세그먼트(hard segment)를 조성하는 것으로 알려져 있다.13,14 따라서
CNC-APS 충전제의 보강 효과가 순수 CNC보다 우수할 것으로 기대된다.
본 연구에서는, 재생이
가능하면서 환경 친화적인 바이오 소재 적용을 확대하기 위하여 대두유를 기반으로 한 바이오 폴리올(B-2466, Mitsui Chemical
& SKC Poyurethanes Inc., MCNS사) 30%와 석유계 poly(tetrahydrofuran)(PTHF) 70%를 혼합하여
실험을 진행하였다. 이 비율에 관한 연구는 본 연구실에서 이미 발표된 논문에 상세히 기술하였다.15 본 연구에서는 CNC-APS 충전제의 보강 성능을 연구하기 위하여, -NCO/OH 몰 비율이 2.2:1이 되도록 폴리올을
과량의 MDI와 반응시켜 양 쪽 말단에 -NCO기를 가지는 예비중합체(prepolymer)를 제조하였다. 그 후, 사슬연장제인 1,4-butanediol (1,4-BD)을 -NCO/OH 비가 1.1:1이 되도록
첨가한 후, 열 경화하여 PU 중합체를 제조하였다. 또한 순수 CNC와 CNC-APS를 충전제로 사용하여 PU 나노복합체를 제조한 후, 제조된
PU 나노복합체의 열적 특성, 기계적 물성 및 충전제의 분산 상태를 연구하였다. Scheme 1에 CNC 표면 개질에 관한 모식도를 나타내었다.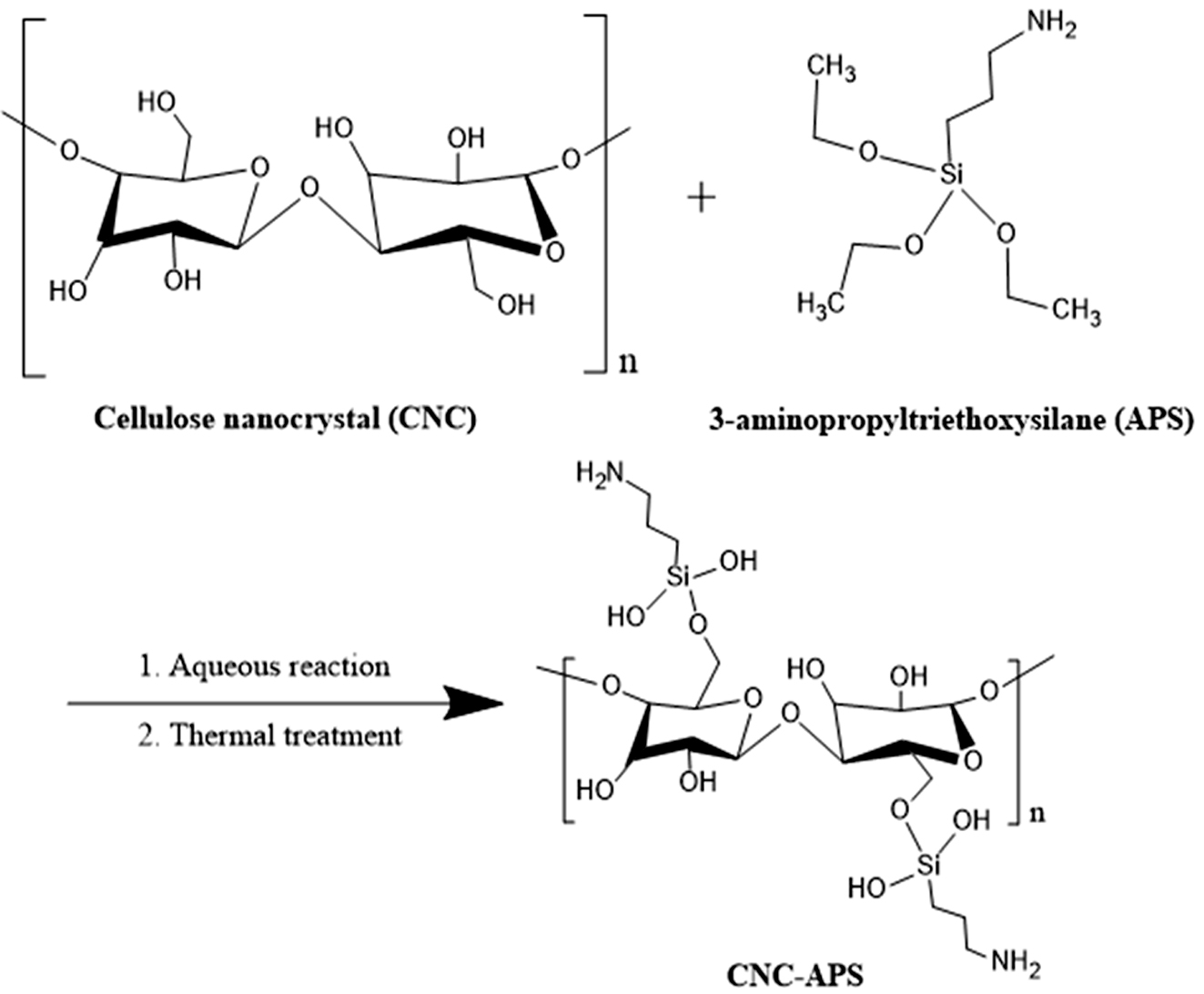
Scheme 1. Scheme for the surface modification of CNC with MPTMS by silanization.
시약 및 재료. Cellulose nanocrystal(CNC, 평균
입자 직경 7.5 nm, 길이 150 nm, CelluForce),
microcrystalline cellulose (Avicel® PH-101, 평균 입자 직경 50 μm, Sigma-Aldrich),
3-aminopropyltriethoxysilane(APS, 99%, Sigma-Aldrich), 탈이온수(deionized water,
MR-RU890, 18.2 MW-cm, Mirae Sci. Corp.,
Korea), ethanol(99.9%, DUKSAN) 및 acetic acid (99.9%, DUKSAN)를 사용하여 CNC 표면개질
실험을 진행하였다. PU 나노복합체 제조에 사용된 poly(tetrahydrofuran) (PTHF, Mw = ~1000 g/mol, 107~118 OH
value, Sigma-Aldrich), 4,4'-methylenebis(phenyl isocyanate)(MDI,
Sigma-Aldrich), 1,4-butanediol(1,4-BD, DUKSAN) 및 dimethyl-formamide(DMF,
Daejung)는 시약급을 구입하여 별도의 정제 없이 그대로 사용하였다. 또한 FTIR 측정용 potassium bromide (KBr, FTIR grade,
Sigma-Aldrich)를 사용하였으며, bio-polyol (B-2466, Mw = ~2400 g/mol, 66 OH value, from soybean
oil)은 MCNS(Mitsui
Chemicals & SKC Polyurethanes Inc.)사로부터 제공받아 사용하였다.
APS에 의한 CNC 표면 개질 반응.16 CNC 표면 개질 반응에 미치는 탈이온수의 pH 영향을
연구하기 위하여 선 가수분해 수용액의 pH를 4.0과 10.5로 조절하였다. pH 4.0 수용액 제조는 acetic acid를 한 방울씩 적하하며
조절하였다. 또한 pH 10.5 수용액 제조 시에는 APS가 아미노기를 가지는 염기성 물질이기 때문에 별다른 pH 조절 없이 탈이온수에 투입하였다.
500 mL 반응기에 탈이온수 300 mL와 APS를 전체 탈이온수 대비 9.0 w/v%(27.0
g) 넣고 pH를 4.0 혹은 10.5로 조절한 후, 기계식 교반기(WISESTIR®, HS-100D)를 사용하여 200 rpm으로 30분 동안 교반하면서 선 가수분해시켰다.
그 후, CNC를 탈이온수 대비
1.0 w/v%(3.0 g) 투입한 후 상온에서 기계식 교반기를 사용하여
200 rpm으로 2시간 반응시켰다.
반응 후 원심분리기(Hettich®, ROTOFIX 32A)를 이용하여 4000 rpm에서 20분 동안 분리시킨 후, 상등액을 버리고
침전된 CNC를 탈이온수를 사용하여 재분산시켰다. 그 후 원심분리기를 사용하여 4000 rpm에서 20분 동안 분리시킨 후, 상등액을 제거하고
남은 CNC는 유리 페트리 디쉬(petri dish)에 옮겨 담아 질소로 3회 치환한 진공오븐(JEOTECH, OV-12)에서 105 oC로 상승시켜 2시간 동안 열처리를 진행하였다.
진공오븐의 온도를
25 oC까지 떨어뜨린 후, CNC와 반응하지 않고 흡착되어 있는 실란을 제거하기 위하여 에탄올(ethanol)에
분산시킨 후 원심분리기를 이용하여 4000 rpm에서 20분 동안 분리하는 과정을 2회 반복하였다. 그 후, 탈이온수를 사용하여 위와 같은 과정을
1회 더 진행하였다. CNC의 수분을 증발시키기 위해 동결 건조 전용 600 mL 비이커(ilShin BioBase,
FB0600)에 CNC 수용액 200 mL를 담아 동결건조기(ilShin BioBase, TFD 5503)로 4일 동안 건조를 진행하였다. 건조된
CNC-APS는 시료에 수분이 흡착되는 것을 방지하기 위하여 데시케이터(desiccator)에 저장하면서 사용하였다.
CNC 및 CNC-APS를 충전제로 사용한 예비중합체 합성.15 50 mL 바이알(vial)에 DMF 20.0 g을 투입하고 CNC 혹은 APS와 반응시켜 표면에 아미노기를
도입한 CNC-APS를 고분자 대비 함량별(1.0, 3.0, 및 5.0 wt%)로 투입한 후, bath형 초음파기(KUDO,
JAC-5020)를 사용하여 상온에서 820 W, 60 Hz로 30분 동안 선 분산을 진행하였다. 그 후 바이알의 용액을 250 mL 반응기에 넣고, DMF 18.26 g, PTHF 7.0 g (0.007 mol) 및 바이오 폴리올 3.0 g(0.0013 mol)을 투입한 후, 60 oC의 맨틀(DAIHAN, WHM12032)을 사용하여 기계식 교반기로 200 rpm에서 2시간 동안 교반하면서
분산시켰다. 이 때, 반응기의 연결부위는 실리콘 그리스를 사용하여 수분을 차단하였다. 그 후, MDI 4.57 g(0.018 mol)을 투입한 후 60 oC에서 기계식 교반기로 200 rpm으로 교반하면서 2시간 동안 반응시켜 예비중합체를 합성하였다.
PU 및 PU 나노복합체 합성. 위 반응으로 예비중합체를 합성한 후 반응기를 맨틀에서
꺼내어 온도를 상온으로 서서히 식힌 후, 사슬 연장제인 1.4-BD를 0.70 g(0.0078 mol) 투입하여 150 rpm으로 1시간 동안 교반하면서 반응시켰다. 그 후
PFA(perfluoroalkoxy) 페트리디쉬(Ø=11 cm)에 두께 약 2.0 mm 높이로 부어 진공오븐에서 밤새 상온 건조 및 탈포
과정을 진행하였다. 그 후, 100 oC의 오븐에서 6시간
용매 제거 및 열 경화를 진행하여 PU 및 PU 나노복합체를 제조하였다.
인장강도 시편제작. 위 방법으로 제조된 PU 및 PU 나노복합체를
ASTM D 1708 규격의 시편 절단기를 사용하여 인장강도 시험용 시편을 제조하였다.
분석. APS를 사용하여 표면 개질을 진행한 CNC의 아미노기
도입 유무를 분석하기 위하여 Fourier transform infrared spectroscopy(FTIR, ThermoFisher Scientific, Nicolet is50),
elemental analysis(EA, Thermo Scientific, FLASH EA1112), X-ray photoelectron
spectroscopy(XPS, ThermoFisher Scientific, K-ALPHA) 및 고체상태 29Si NMR(Bruker, AVANCE III HD
400)를 사용하였다.
FTIR 분석은
diffuse reflectance infrared Fourier transform spectroscopy(DRIFTS-FTIR)법을 이용하였다.
순수 KBr을 갈아서 background로 사용하고, KBr과 순수 시료를 95:5 wt%로 혼합한 후, 갈아서 4000~400 cm-1 파장에서 4 cm-1 분해능으로 64회 스캔(scan)하여
측정하였다. DRIFTS-FTIR 스펙트럼은 Kubelka-Munk function(K-M)으로 변환하여 정량적 분석을 진행하였다. 또한 시료
전체의 탄소 및 질소 성분의 양은 EA(elemental analysis) 분석을 실행하였으며, 표면 10 nm 깊이의 화학 성분은 XPS를 사용하여 시료를
50 eV pass energy에서 0.82 eV 분해능으로 10회 스캔하여 분석하였다.
pH에 따라 개질된 CNC에 접목된 APS의 구조 변화를
측정하기 위하여 고체상태 29Si NMR(Bruker, AVANCE III HD 400)를 사용하여 CP/MAS(cross
polarization/magic angle spinning) 법으로 회전속도 400 MHz로 측정하였다.
또한 충전제를 사용하여
제조한 PU 나노복합체는 FTIR, TGA(Scinco, TGA N-1000), UTM(Qmesys, QM100SE), DMA(TA, DMA Q800) 및 field
emission scanning electron microscope(FE-SEM, Hitachi, SU8220) 분석을 실시하였다.
FTIR 분석은 다이아몬드(diamond)
결정을 사용한 전반사법(attenuated total reflection, ATR) accessory를 사용하여 4000~400 cm-1에서 4 cm-1 분해능으로 32회 스캔하여
측정하였다.
TGA 분석은 질소분위기
하에서 30 oC에서 800 oC까지 10 oC/min의 속도로 온도를 올리면서 시료의 무게 변화를 측정하였다. UTM 분석은 ASTM D 1708 규격으로
시편을 제조하여 5 mm/min의 속도로 측정하였다. 시편을 5개씩 제조하여
인장강도, 파단연신율 및 탄성률의 평균값과 표준편차를 구하였다. DMA 분석은 10×40×1 mm 규격으로 시편을 제조하여 1 Hz의 측정 주파수에서 -120~150 oC 범위에서 3 oC/min의 속도로
승온시키며 저장탄성률, 손실탄성률 및 tan δ 값을 측정하였다. FE-SEM 분석은 PU 나노복합체의 시편을 액체질소로 냉각한 후, 파단 시켜
약 10 nm 두께의 Pt로 코팅하여 5 kV의 가속 전압에서
분석하였다.
APS에 의한 CNC 표면 개질 반응 분석. APS를 사용하여 표면 개질을 진행한 CNC의 아미노기
도입을 확인하기 위하여 DRIFTS-FTIR법을 사용하여 스펙트럼을 측정하였다. 그 결과는 Figure 1에 나타내었다.
Figure 1에서
(a)는 순수 CNC, (b)는 pH 4.0 수용액에서 APS로 개질된 CNC-APS, (c)는 pH 10.5 수용액에서 개질된 CNC-APS
스펙트럼을 각각 나타내고 있다.
Figure 1(b)와
1(c) 스펙트럼에서는 1(a)에서는 관찰되지 않던 피크가 1560과 1596 cm-1에서 각각 새롭게 나타나는
것을 확인하였다. Figure 1(b) 스펙트럼의 1560 cm-1에서 새로 나타나는 피크는 -NH3+기의
굽힘(bending) 피크로 판단된다. 아민기의 질소는 고립 전자쌍(lone pair)을 가지고 있으며, 아민기의 고립전자쌍과 pH 조절에 사용된 acetic acid의 양성자와
반응하여 -NH3+기가 생성된 것으로 판단된다.17,18 또한 Figure 1(c)에서는
일차 아미노기(-NH2기)의 굽힘 진동 피크가 1596 cm-1에서 나타난 것으로 판단된다.19
이로 인해 CNC 표면에 APS의 실란화 반응에 의하여 아미노기가 성공적으로 도입되었음을 확인하였다.
APS를 사용하여
CNC 표면에 도입된 N 원소 함량을 측정하기 위하여 EA 분석을 수행하여 Table 1에 나타내었다.
EA 분석 결과,
pH 4.0 수용액에서 개질을 진행한 경우 C, H, O, N 및 S의 함량이 각각 43.23, 6.31, 48.22, 1.81 및 0.43%이고,
pH 10.5에서 개질을 진행한 경우 42.32, 6.45, 46.76, 4.16 및 0.32%로 각각 측정되었다. 위 결과들을 비교하여 보면,
순수 CNC에서는 N의 함량이 0%이지만 pH 10.5에서 개질된 CNC-APS의 N 함량은 4.16%로 pH 4.0에서 개질된 CNC-APS의
1.81%보다 높았다. 따라서 pH 10.5에서 개질된 CNC-APS에 더 많은 -NH2기가 접목된 것으로 판단된다. S의 함량은 순수 CNC, pH 4.0에서 개질된
CNC-APS, pH 10.5에서 개질된 CNC-APS에서 각각 0.61, 0.43 및 0.31%로 나타났다. 이러한 S의 존재는 CNC 제조
시, 셀룰로오스 섬유에 황산을 가하면 CNC 표면에 있는 -OH기의 일부가 황산과 에스테르화 반응(esterification)을
일으켜 음전하를 띠는 sulfate ester group(-SO3-)로 변화하기 때문으로 판단된다.20
APS는 1분자 당
1개의 N 원자에 대하여 3개의 C 원자를 가지는 구조로써, 셀로비오스(cellobiose) 한 분자에 2개씩 존재하는 1차 -OH기 중에서(1차
-OH기가 2차 -OH기보다 반응성이 큰 것으로 알려져 있음), 하나 및 두 개의 1차 -OH기가 APS와
실란화 반응할 경우 N/C 원소 비율은 각각 0.07과 0.11이 된다. pH 4.0과 10.5에서 개질을 진행한 CNC-APS의 C와 N 원자량을
고려하여 계산한 N/C 값은 각각 0.04와 0.08로 셀로비오스 한 분자 당 약 0.57개 및 1.14개의 APS가 반응하는 것으로 판단된다.
그러나 이 계산은 APS가 셀로비오스 표면에 위치한 1차 -OH기와 반응하여 단일 층을 형성한다는 가정에서 계산한
결과이다. 실란화제가 고체 표면의 -OH기와 실란화 반응을 할 때, 단일 층뿐만 아니라 멀티 층(multilayer)을 형성할 수
있기 때문에, 셀로비오스 표면의 1차 -OH기 중에서 APS와 결합하는 수는 더 적은 것으로 판단된다.
CNC 표면에 도입된
APS를 확인하기 위하여 XPS를 사용하여 표면 10 nm 깊이를 분석한 survey scan 결과를 Figure 2에 나타내었다.
Figure 2의
(a), (b) 및 (c) 스펙트럼에서는 공통으로 C1s, O1s 및 S2p의 결합에너지가
286.1, 532.4 및 169.0 eV에서 각각 나타났다. 순수 CNC 스펙트럼에서는 보이지 않던 N1s, Si2s
및 Si2p 피크가 (b)와 (c)에서 각각 400.1, 101.6 및 152.8 eV에서 나타났다. 이로 인하여 CNC 표면에
APS가 도입되었음을 알 수 있었다. Figure 2의 스펙트럼 결과에서 각각의 원소함량을 Table 2에서 나타내었다.
Table 2의
XPS 분석 결과에서 보면, pH 4.0과 10.5에서 APS 개질을 진행한 CNC-APS의 N/C 원소 비율은 각각 0.07과 0.10으로
EA 분석 결과인 0.04와 0.08보다 더 높은 것을 알 수 있었다. 따라서 APS가 CNC 표면에 더 많이 접목된 것으로 판단된다.
CNC 표면에
APS가 도입된 후의 구조 변화를 분석하기 위하여 29Si NMR 분석을 진행하여 그 결과를 Figure 3에 나타내었다.
Figure 3(a)와
3(b)는 각각 pH 4.0 및 10.5 수용액에서 APS 개질을 진행한 CNC의 고체상태 NMR 스펙트럼이다. Figure 3(a)와
3(b) 모두 APS의 Si가 도입되어, -49.8, -59.6 및 -68.5 ppm에서 각각 T1
(SiO(OH)2R'), T2 (SiO2(OH)R') 및 T3
(SiO3R')의 피크들을 확인할 수 있었다.21 두 스펙트럼의 면적 변화를 정량적으로
비교하기 위하여 NMR 기기에 설치되어 있는 소프트웨어인 Topspin을 사용하여 Deconvolution하여, T1, T2
및 T3의 면적 비율을 측정하였다. 측정 결과 Figure 3(a)는 각각 27.3, 46.9 및 25.8%이고,
3(b)는 7.3, 44.7 및 48.0%로 나타났다. Figure 3(a)와 3(b)를 비교하면,
3(a)의 T1 면적이 3(b)보다 크고 T3 면적은 작은
것을 알 수 있다. 이는 산성 수용액에서는 가수분해반응으로 -SiOH 생성이 유리하고 염기성 수용액에서는 -SiOH기의 자기축합반응(self-condensation)으로
올리고머 생성이 유리하기 때문으로 판단된다. 즉, 산성에서 개질된 CNC-APS는 T1 구조의 생성이 상대적으로 많고 염기성에서
개질된 CNC-APS는 T3 구조의 생성이 상대적으로 더 많이 일어나는 것으로 판단된다.22,23
예비중합체 합성. PTHF와 바이오 폴리올을 7:3의 비율로 혼합한 후
MDI와 반응시켜 예비중합체 제조 시, 반응이 종료됨을 확인하기 위하여 30분 간격으로 시료를 샘플링(sampling)하여 FTIR-ATR법으로
측정하였다. 측정한 스펙트럼을 투과법으로 측정한 스펙트럼으로 변환하기 위하여 ATR correction을 수행하여 Figure 4에 나타내었다.
Figure 4를 보면
2268, 1731 및 1600 cm-1에서 각각 MDI의 미반응
-NCO 피크, 우레탄의 C=O 피크 및 MDI 벤젠 링의 C=C 피크를 확인할 수 있었다.24 반응시간이 증가할수록
MDI의 -NCO기가 폴리올의 -OH기와 반응하여 우레탄 결합을 형성하기 때문에 2268 cm-1 피크의 면적이 점점 감소하고,
-NCO기가 모두 반응하여 반응이
종결되면 -NCO 피크가 사라질 것으로 판단된다.25 따라서 1600 cm-1에서 나타나는 벤젠 링의
C=C 피크를 기준으로 2268 cm-1의 미반응 -NCO 피크의 면적
변화를 식 (1)을 사용하여 면적 비를 계산하여 반응 정도를 정량적으로 분석하였다.
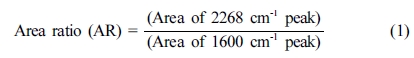
반응 시간이 0에서
2시간까지 30분 간격으로 증가함에 따라 2268 cm-1의 피크 면적은 9.92, 5.76, 2.33, 4.74 및 4.65로 줄어들고,
1600 cm-1의 피크 면적은 1.53,
1.54, 1.48, 1.57 및 1.58로 나타났다. 이 값들을 식 (1)을 이용하여 계산한 AR 값은 시간이 지남에 따라 6.48,
3.74, 3.60, 3.02 및 2.94로 점점 감소하였다.
위 결과를 사용하여
계산하면 2시간 반응 후에는 MDI의 54.6%가 반응하고 45.4%가 미반응 상태로 남아있음을 알 수 있었다. 폴리올의 -OH기와 MDI의
-NCO기는 서로 1:1로 반응을 진행하는 것으로 알려져 있으나, 반응에 투입된 MDI가 실험 중에 의도하지 않게 도입되는 수분과 반응할
수 있다.26 따라서 이러한 점을 보완하기 위하여 NCO/OH의 비를 2.2:1로 MDI를 넣어 실험을 진행하였다.
과량으로 투입된
MDI의 -NCO기는 수분과 반응하면 -NH2기를 생성한 후, MDI의 -NCO기와 반응하여
우레아 결합이 생성 가능하다.27,28 이러한 반응으로 생성된 우레아의 free C=O 피크가 1712 cm-1에서 shoulder로 나타나는
것을 확인할 수 있었다. 1,4-BD를 사용해 1시간 동안 사슬연장 반응을 진행한 PU 나노복합체의 스펙트럼에선 2268 cm-1의 -NCO 피크가 완전히
사라진 것으로 보아 중합 반응이 완료되었음을 확인하였다.29
충전제를 사용한 PU 나노복합체 열적 안정성 분석. 1.0 wt%의 순수 CNC와 pH 4.0과 10.5 수용액에서
개질한 CNC-APS를 충전제로 각각 사용한 PU 나노복합체들의 열적 분해 거동을 TGA를 사용하여 분석하여 Figure 5에 나타내었다.
Figure 5(a)와
5(b)에 PU 나노복합체들의 TGA 결과와 DTG (derivative thermogravimetry) 결과를 각각 나타내었다. Figure 5(a)를
보면, PU 나노복합체들의 분해가 순수 PU 중합체와 동일하게 2단계로 진행되며, 열분해 시작 온도는 모두 334 oC로 측정되었다. Figure 5(b)의 DTG 곡선에서 순수 PU 중합체의 최대 분해 속도 온도는 360과 438 oC로 나타났다. 또한 CNC와 pH 4.0과 10.5에서 각각 개질한 CNC-APS를 충전제로 사용한 PU 나노복합체의 최대
분해 속도 온도는 각각 374와 443 oC, 371과
445 oC 및 372와 446 oC로 각각 나타났다.
이러한 결과에서 충전제를 사용한 PU 나노복합체의 첫 번째 최대 분해 속도가 나타나는 온도가 순수 PU보다 약 10 oC 이상 높은 것을 확인하였다. 이는 MDI의 -NCO기와 CNC 표면의 -OH 혹은 -NH2
기가 계면에서 우레탄 혹은 우레아 결합을 형성하여 열적 안정성이 높아진 것으로 판단된다. 그러나 CNC 충전제와 CNC-APS 충전제의 열분해
온도 차이는 2~3 oC로 큰 차이가 없는
것을 알 수 있었다.
800 oC에서 잔류량의 경우, 순수 PU 중합체는 4.88%, CNC와 pH 4.0 및 10.5의 수용액에서 개질한
CNC-APS를 충전제로 사용한 PU 나노복합체는 각각 6.35, 8.19 및 8.59%로 측정되었다. 셀룰로오스 충전제가 고분자 물질에 충전제로
사용될 경우 잔류랑이 증가되는데, 이는 CNC와 CNC-APS에 존재하는 탄소와 수소 원소 이외의 O, N 및 Si와 같은 헤테로원자(heteroatom)로
인하여 연소 과정에서 char가 생성되어 표면층을 형성하면서 분해과정을 억제시키기 때문으로 알려져 있다.30 특히,
CNC-APS를 충전제로 사용한 PU 나노복합체의 경우에는 CNC 표면에 도입된 APS의 N과 Si 성분에 의해 잔류량이 많이 남는 것으로 판단된다.
충전제를 사용한 PU 나노복합체 기계적 물성 분석. 충전제의 종류에 따라 PU 나노복합체의 기계적 물성에
미치는 영향을 연구하기 위하여 microcrystalline cellulose(MCC), CNC, CNC-APS 1.0 wt%를 각각 충전제로
사용하여 PU 나노복합체를 제조 후 UTM으로 측정하여 응력-변형률 곡선을 Figure 6에 나타내었다.
Figure 6을 보면,
충전제를 사용하지 않은 순수 PU의 인장강도, 연신율 및 탄성률은 4.66±0.16 MPa, 392.64±6.00% 및 21.76±1.14 MPa로 측정되었다. MCC, CNC,
CNC-APS(pH 4.0) 및 CNC-APS(pH 10.5)를 각각 1.0 wt% 충전제로 사용한 PU 나노복합체의 인장강도는 각각 5.41±0.28, 6.82±0.36, 7.68±0.63
및 8.89±0.36 MPa, 탄성률은 25.96±2.25, 30.48± 4.18, 44.51±2.60 및 50.11±3.11 MPa, 연신율은 197.86±6.23, 287.71±5.18,
628.39±8.42 및 615.75±7.55%로 나타났음을 확인하였다.
MCC와 CNC 충전제를
사용하여 제작한 PU는 순수 PU보다 높은 인장강도와 탄성률, 그리고 낮은 연신율을 나타내었다. 셀룰로오스는 축 방향 탄성계수가 134 GPa로
알려진 보강 충전제로, 이러한 현상은 보강 충전제를 사용하여 제조된 복합체에서 일반적으로 나타나는 현상이다.31
충전제의 크기, 표면적,
형상 및 표면 활성도가 복합체의 물성에 영향을 미치는 것으로 알려져 있다. CNC를 충전제로 사용한 PU 나노복합체의
연신율과 탄성률이 MCC를 충전제로 사용하였을 때보다 높은 것을 알 수 있다. 본 실험에 사용된 MCC의 종횡비(aspect ratio, f )는 2~4로 알려져 있고, CNC의 종횡비는 20으로 알려져
있다.32,33 또한 MCC의 비표면적(specific surface area)은 0.08 m2/g이고,34
CNC의 비표면적은 400 m2/g으로 알려져 있다.35
따라서 CNC가 MCC보다 높은 종횡비와 비표면적을 가지고 있어 PU 매트릭스와 상호작용할 수 있는 계면이 크기 때문에 고분자와 더 강한 네트워크를
형성하여 기계적 물성이 높은 것으로 판단된다.36 또한 순수 CNC를 충전제로 사용한 PU보다 APS를 사용하여 개질한 CNC-APS를 충전제로
사용한 PU의 인장강도, 연신율 및 탄성률이 더 높은 것을 확인하였다. 이는 CNC-APS의 -NH2기와 MDI의 -NCO기가 계면에서
우레탄 결합보다 더 강한 우레아 결합을 형성하기 때문으로 판단된다. 또한 Figure 6(d)와 6(e) 그래프를
보면 pH 4.0보다 10.5에서 개질한 CNC를 충전제로 사용한 PU 나노복합체의 기계적 물성이 더 우수함을 알 수 있다. 이는
CNC-APS의 FTIR 및 XPS 분석 결과에서와 같이 pH 10.5에서 APS로 개질된 CNC-APS에 pH 4.0에서 개질된
CNC-APS보다 더 많은 -NH2기가 CNC 표면에 도입된 영향으로 판단된다.
또한 Figure 6(d)와 6(e)의 곡선을 순수 CNC를 포함하는 6(c) 곡선과 비교하면 인장강도와 연신율이 동시에 증가하는
것을 알 수 있었다. 이는 CNC 표면에 접목된 -NH2기가 PU의 유연한 부분(soft segment)보다 hard
microdomain을 우선적으로 강화하여 유연한 부분의 움직임이 제한을 받지 않아 연신율과 탄성률이 동시에 증가되는 것으로 판단된다.37
pH 10.5에서
개질한 CNC-APS 충전제를 사용하여 충전제의 함량이 PU 나노복합체의 기계적 물성에 미치는 영향을 알아보기 위하여 1.0, 3.0 및
5.0 wt%를 투입하여 제조한 PU 나노복합체를 UTM으로 측정한 응력-변형률 곡선을 Figure 7에 나타내었다.
Figure 7을 보면
CNC-APS의 투입량을 0, 1.0, 3.0 및 5.0 wt%로 증가하면 인장강도는 4.66±0.16, 7.68±0.63, 10.48± 0.36 및 9.78±0.27 MPa, 탄성률은 21.76±1.17, 44.51±2.60, 56.69±2.27
및 60.18±1.64 MPa, 연신율은 392.64±6.00, 628.39±8.42, 338.68±10.43 및 324.86±9.77%로
나타났음을 확인하였다. 이 결과들을 순수 PU 중합체의 응력-변형률 곡선인 Figure 7(a)와 비교하면
인장강도는 1.65, 2.25 및 2.10배 증가하고, 탄성률은 2.05, 2.61 및 2.77배 증가하였다. 탄성률이 3 wt%까지는 급격히 증가하다가 5.0 wt%까지는 완만히 증가하는 이유는 충전제의 함량이
5.0 wt%로 증가하면서 충전제들이 응집체(agglomerate)를
형성하여 고분자와의 계면 결합력이 감소하기 때문으로 판단된다.38 연신율은 충전제의 함량이 1.0 wt%일 때 순수 PU 중합체보다 1.60배 증가하고
3.0 및 5.0 wt%일 때 0.86, 0.83배로 감소하였다. 이는
충전제의 함량이 높아질수록 충전제의 분산성이 감소하여 응집체를 형성하기 때문에 연신율이 점차 낮아지는 것으로 판단된다.39
충전제를 사용한 PU 나노복합체 동역학적 거동 분석. 순수 PU 중합체와 CNC 1.0 wt%, CNC-APS(pH 10.5) 1.0,
3.0 및 5.0 wt%를 각각 충전제로 사용하여 제조한 PU 나노복합체의
점탄성 거동을 DMA를 사용하여 측정한 결과를 Figure 8에 나타내었다.
Figure 8(a)에
PU 중합체와 나노복합체의 저장탄성률(E′)을 나타내었다. 순수 PU 중합체의 유리영역인 -70 oC에서의 E′는 865 MPa, 고무영역인 20 oC에서의 E′는 17 MPa로 측정되었다.
CNC 1.0
wt%, CNC-APS 1.0, 3.0 및 5.0 wt%를 충전제로 사용한 PU 나노복합체의 -70 oC에서의 유리영역의 E′는 1187, 1331, 1173 및 1290 MPa로 측정되었고,
20 oC에서의 고무영역의 E′는 21, 25, 27 및 30 MPa로 측정되었다. 이를 순수 PU 중합체와 비교하면
유리영역의 E′는 각각 1.37, 1.54, 1.36 및 1.49배로 나타났으며 고무영역의 E′는 1.24,
1.47, 1.59 및 1.76배로 나타났다. 순수 PU 중합체보다 충전제를 함유한 PU 나노복합체의 E′가 더 높게 나타나는 것을
알 수 있었다. 이는 CNC 혹은 CNC-APS 입자와 PU 고분자가 계면에서 우레탄 결합과 우레아 결합을 형성하면서 보강 효과가 증가하기 때문으로
판단된다.
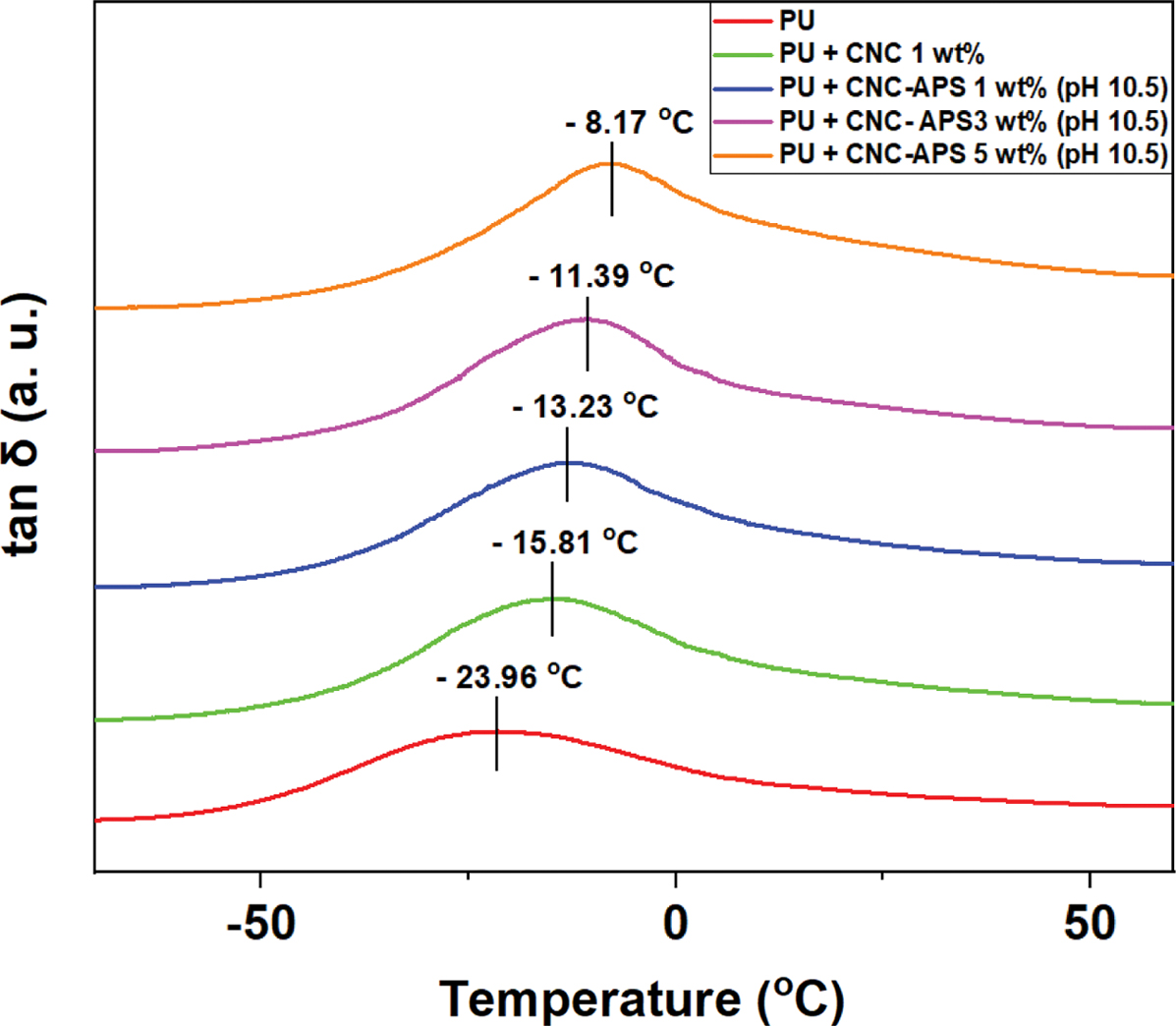
Figure 8(b)는
PU 중합체와 나노복합체의 tan δ 값을 나타낸 것으로, tan δ 값은 손실탄성률(E″)과 E'의 비(E″/E′)이며,
tan δ의 피크가 나타나는 온도는 고분자의 Tg와 연관이 있는 것으로 알려져 있다. 순수 PU 중합체,
CNC 1.0 wt%, CNC-APS 1.0, 3.0 및 5.0 wt%를 충전제로 사용한 PU 나노복합체의 Tg
값은 각각 -23.96, -15.81, -13.23, -11.39 및 -8.17 oC로 점점 증가하는 것을 알 수 있었다.
MDI의 -NCO기는 폴리올과
CNC 표면의 -OH기와 만나면 우레탄 결합을 생성하고 CNC-APS의 -NH2기와 만나면 우레아 결합을 생성할
수 있다. -NCO기와 1차 -OH기 및 2차 -OH기와의 상대반응성(relative
reactivity)은 각각 2.5와 0.7이고, -NH2기와의 상대반응성은 2500으로 알려져 있다.40 따라서 MDI의 -NCO기는
CNC-APS의 -NH2기와 상대적으로 빨리 반응하여 우레탄 결합보다 강한 우레아 결합을 생성할 수 있다.
이로 인해 고분자 사슬의 거동에 더 크게 제한을 받아 순수 CNC보다 CNC-APS의 Tg 값이 높게 측정되고,
CNC-APS의 함량이 높아질수록 Tg 값이 높아지는 것으로 판단된다.
PU 나노복합체 충전제 분산상태 및 계면 결합력 분석. PU 나노복합체의 계면에서의 결합정도 및 충전제 분산
상태를 더 자세히 분석하기 위하여 FE-SEM을 사용하여 시료의 파단면을 50000배로 측정하여 Figure 9에 나타내었다.
Figure 9(a)는
순수 CNC 1.0 wt%를 투입하여 제조한 PU 나노복합체, 9(b),
9(c) 및 9(d)는 CNC-APS (pH 10.5)를 각각 1.0, 3.0 및 5.0 wt% 투입하여 제조한 PU 나노복합체의 파단면이다.
FE-SEM에 설치되어 있는 PC-CHEM 소프트웨어를 사용하여 막대 모양의 입자 10개의 크기(길이와 직경)를 측정하여 평균값과 표준 편차를
계산하였다. Figure 9(a), 9(b), 9(c) 및 9(d)에서 관찰되는 충전제 입자들의 평균 길이는 각각 154.3±25.53,
144.4±33.3, 147.1±17.2 및 153.56± 14.2 nm로 본 실험에 사용한 CelluForce사의 CNC 평균 길이인 150 nm와 거의 일치한 길이를 나타내었다. 충전제 입자들의
평균 직경은 각각 38.9±8.9, 34.04±5.8, 32.9±3.9 및 52.54±9.2 nm로 CNC 평균 직경인 7.5 nm보다 높아서 응집체를 형성하는 것을 알 수 있었다.
이 평균 직경과 CNC의 평균 직경을 사용하여 응집체 한 개 당의 CNC의 평균 개수를 계산하면, 4~7, 3~5, 3~5 및 5~8 범위로서 순수 CNC와 5.0 wt%의
CNC-APS 충전제를 사용하였을 때 응집체를 더 크게 형성한 것으로 판단된다.
Figure 9(a)에서
친수성인 순수 CNC 충전제들과 소수성인 PU 계면과의 결합력이 약하여 탈결합(debonding)이 일어나서 CNC 입자의 옆면을 용이하게 관찰할
수 있었다. 그러나 Figure 9(b)에서는 CNC-APS의 아미노기와 MDI의 -NCO기가 우레아 결합을 형성하여 계면 결합력이 증가하여
CNC-APS 입자들이 PU 나노복합체에 묻혀있는 상태로 존재하는 것을 관찰할 수 있었다. 즉, CNC-APS를 충전제를 사용하여 제조한 PU
나노복합체가 순수 CNC를 사용하여 제조한 PU 나노복합체보다 분산성이 우수하고 계면 결합력이 우수한 것을 알 수 있었다.
또한 Figure 9(b),
9(c) 및 9(d)에서 CNC-APS 충전제의 함량이 1.0, 3.0 및 5.0 wt%로 증가할수록 CNC-APS의 분산성이 감소하여
응집체를 더 크게 형성하는 것을 관찰할 수 있었다. 특히 Figure 9(d)에서는 응집체가 크게 형성된 것으로 보여주고 있다.
Figure 9(d)와 같은 응집체가 형성되면, 충전제 입자와 고분자 사이에는 접촉할 수 있는 표면적이 줄어들어서 계면
결합력이 감소하고, 응집체 입자 사이의 약한 결합력 때문에 응집체 내부에 공극이 생기는 것을 관찰할 수 있었다.

|
Figure 1 DRIFTS-FTIR spectra with different pH values: (a) pristine CNC; (b) CNC modified with 9.0 w/v% APS at pH 4.0; (c) CNC modi fi ed wi th 9.0 w/v% APS at pH 10.5. |

|
Figure 2 XPS survey spectra of (a) pristine CNC; (b) CNC modified with 9 .0 w/v% APS at pH 4.0; (c) CNC modi fi ed wi th 9 .0 w/v% APS at pH 10.5. |

|
Figure 3 Solid state 29Si NMR spectra of (a) CNC modified with 9.0 w/v% APS at pH 4.0; (b) CNC modified with 9.0 w/v% APS at pH 10.5. |

|
Figure 4 FTIR-ATR spectra of PU with different pre-polymerization time. |

|
Figure 5 TGA weight loss curves and DTG curves of (a) PU; (b) PU with 1.0 wt% CNC; (c) PU with 1.0 wt% CNC-APS (pH 4.0); (d) PU wi th 1.0 wt% CNC-APS (pH 10.5). |

|
Figure 6 Stress-strain curves of (a) PU; (b) PU with 1.0 wt% MCC; (c) PU wi th 1 .0 wt% CNC; (d) PU with 1 .0 wt% CNC-APS (pH 4.0); (e) PU with 1.0 wt% CNC-APS (pH 10.5). |

|
Figure 7 Stress-strain curves of (a) PU; (b) PU with 1.0 wt% CNCAPS (pH 10.5); (c) PU with 3.0 wt% CNC-APS (pH 10.5); (d) PU wi th 5.0 wt% CNC-APS (pH 10.5). |

|
Figure 8 DMA curves of PU and PU composites with different fillers: (a) storage modulus curves; (b) tan δ curves. |

|
Figure 9 FE-SEM images of (a) PU with 1.0 wt% CNC; (b) PU with 1.0 wt% CNC-APS (pH 10.5); (c) PU with 3.0 wt% CNC-APS (pH 10.5); (d) PU wi th 5.0 wt% CNC-APS (pH 10.5). |

|
Table 2 Elemental Surface Composition of Pristine and APS Modified CNCs from XPS Survey Spectra |

본 연구에서는 실란
커플링제인 APS를 사용하여 CNC 표면에 아미노기가 도입된 CNC-APS 충전제 첨가가 친환경 PU 나노복합체의 물성에 미치는 영향에 대한 연구를
진행하였다.
APS를 사용하여
CNC 표면 개질을 진행한 후 아미노기가 도입되었는지 확인하기 위해 FTIR을 사용하여 분석한 결과, 순수 CNC에서는 보이지 않던 N-H 피크가
개질된 CNC에서는 나타나는 것을 확인하였다. 또한 EA 및 XPS를 사용하여 순수 CNC에서는 관찰되지 않던 N과 Si 원소가 개질된
CNC-APS에서는 관찰되는 것을 확인하였다. 이로 인해 CNC 표면에 아미노기가 도입되었음을 알 수 있었다.
순수 PU 중합체
및 CNC와 CNC-APS 1.0 wt%를 충전제로 사용한 PU 나노복합체의 열적 특성을
TGA를 사용하여 분석한 결과, 충전제가 투입됨으로써 열적 안정성이 높아지는 것을 확인하였다. 또한 CNC 충전제보다 CNC-APS 충전제를 사용하여
제조한 PU 나노복합체의 800 oC에서의 잔류량이 더 높은 것을 확인하였다. 이는 APS의 N과 Si 원소가 char를 형성하기 때문으로 판단된다.
UTM 분석 결과,
MCC보다 CNC를 충전제로 사용하였을 때 탄성률이 더 높은 것을 확인하였다. 이는 CNC의 종횡비가 MCC의 종횡비보다 크고 비표면적이 더 크기
때문에 더 우수한 보강효과를 가지는 것으로 판단된다. 또한 CNC보다 CNC-APS를 충전제로 사용하였을 때 탄성률이 더 높고, CNC-APS의
함량이 증가할수록 인장강도와 탄성률이 증가하는 것을 확인하였다. 이는 CNC-APS의 -NH2기와 MDI의 -NCO기가 계면에서
더 강한 우레아 결합을 형성하기 때문에 우레탄 결합을 형성할 수 있는 순수 CNC보다 기계적 물성이 우수한 것으로 판단된다.
또한 CNC 표면에
접목된 -NH2기가 PU의 유연한 부분보다 hard microdomain을 우선적으로 보강하여 유연한 부분의 움직임이 제한을
받지 않아 연신율과 탄성률이 동시에 증가되는 것을 확인하였다.
DMA 분석 결과,
순수 PU 중합체보다 CNC와 CNC-APS를 충전제로 사용하여 제조한 PU 나노복합체의 Tg 값이 더 높아지는
것을 알 수 있었다. 이는 MDI의 -NCO기가 순수 CNC 충전제 표면의 -OH기와 반응하여 우레탄 결합을 생성하거나,
CNC-APS 충전제 표면의 -NH2기와 반응하여 우레아 결합을 형성하여 고분자 사슬의 거동에 제한을 받기
때문으로 판단된다. 또한 CNC-APS의 함량이 증가할수록 PU 나노복합체의 Tg 값이 증가하는 것을 확인하였다.
FE-SEM 분석 결과, 1.0 wt% 충전제 함량에서, CNC-APS 입자들이 순수
CNC 입자보다 응집체를 적게 형성하고 분산이 균일하게 되는 것을 확인하였다. 또한 CNC-APS의 함량이 5.0 wt%가 되면 응집체가 형성되면서
고분자와의 계면 결합력이 약해져 응집체 내부에 공극이 생기는 것을 알 수 있었다.
- 1. Y. J. Jo, S. H. Choi, and E. Y. Lee, Appl. Chem. Eng., 24, 579 (2013).
-
- 2. C. B. Kim, W. J. Seo, O. D. Kwon, and S. -B. Kim, Appl. Chem. Eng., 22, 540 (2011).
- 3. O. J. Kwon, S. R. Yang, D. H. Kim, and J. S. Park, J. Appl. Polym. Sci., 103, 1544 (2007).
-
- 4. R. C. Saxena, D. K. Adhikari, and H. B. Goyal, Renew. Sust. Energ. Rev., 13, 167 (2009).
-
- 5. I. T. Kim, S. H. Joo, and J. S. Oh, J. Adhes. Intf., 20, 116 (2019).
- 6. B. J. Kim, S. Y. Kim, J. K. Cho, S. H. Lee, and H. J. Kim, J. Adhes. Interf., 11, 76 (2010).
- 7. J. H. Jang, G. J. Kwon, J. H. Kim, S. M. Kwon, S. L. Yoon, and N. H. Kim, J. Korean Wood. Sci. Tech., 40, 156 (2012).
-
- 8. B. L. Peng, N. Dhar, H. L. Liu, and K. C. Tam, Can. J. Chem. Eng., 89, 1191 (2011).
-
- 9. V. Khoshkava and M. R. Kamal, Biomacromolecules, 14, 3155 (2013).
-
- 10. Y. S. Nam, J. K. Yu, G. W. Park, H. J. Kim, D. G. Kim, M. Kim, and K. Hyun, Polym. Korea, 43, 567 (2019).
-
- 11. S. Lee and K. Ha, Polym. Korea, 37, 777 (2013).
-
- 12. N. Luo, D.-N. Wang, and S.-K. Ying, Macromolecules, 30, 4405 (1997).
-
- 13. C. Deng, Y. Cui, T. Zhao, M. Tan, H. Huang, and M. Guo, RSC Adv., 4, 24095 (2014).
-
- 14. J. O. B. Asplund, T. Bowden, T. Mathisen, and J. Hilborn, Macromolecules, 39, 4380 (2006).
-
- 15. Y. Lee, J. Lee, S. Park, K.-H. Lim, and K. Ha, Polym. Korea, 43, 652 (2019).
-
- 16. H. Khanjanzadeh, R. Behrooz, N. Bahramifar, W. Gindi-Altmutter, M. Bacher, M. Edler, and T. Griesser, Int. J. Biol. Macromol., 106, 1288 (2018).
-
- 17. B. C. Smith, Spectroscopy, 44, 30 (2019).
- 18. N. B. Colthup, L. H. Daly, and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, New York-London, 1975.
- 19. H. Kargarzadeh, R. M. Sheltami, I. Ahmad, I. Abdullah, and A. Dufresne, Polymer, 56, 346 (2015).
-
- 20. J. Shin, NICE, 34, 500 (2016).
- 21. J. Luo, J. Lannutti, and R. Seghi, J. Adhes. Sci. Technol., 15, 267 (2001).
-
- 22. M. C. B. Salon, G. Gerbaud, M. Abdelmolleh, C. Bruzzese, S. Boufi, and M. N. Belgacem, Magn. Reson. Chem., 45, 473 (2007).
-
- 23. A. Rachini, M. L. Troedec, C. Peyratout, and A. Smith, J. Appl. Polym. Sci., 123, 601 (2012).
-
- 24. Z. Rafiee and V. Keshavarz, Prog. Org. Coat., 86, 190 (2015).
-
- 25. C. S. Wong and K. H. Badri, Mater. Sci. Appl., 3, 78 (2012).
-
- 26. I. S. Cho, S. G. Kang, and S. B. Kim, J. Korean Inst. Gas, 12, 38 (2008).
- 27. K. K. Jena, D. K. Chattopadhyay, and K. V. S. N. Raju, Eur. Polym. J., 43, 1825 (2007).
-
- 28. O. Ø. Knudsen and A. Forsgren, Corrosion Control Through Organic Coatings, CRC Press, Boca raton, p 20 (2017).
-
- 29. M. J. Choi, B. Y. Jeong, J. M. Cheon, K. B. Park, and J. H. Chun, J. Adhes. Interf., 18, 8 (2017).
-
- 30. H. Koga, T. Kitaoka, and A. Isogai, J. Mater. Chem., 21, 9356 (2011).
-
- 31. Q. Wu, M. Henriksson, X. Liu, and L. A. Berglund, Biomacromolecules, 8, 3687 (2007).
-
- 32. D. M. Panaitescu, A. N. Frone, M. Ghiurea, C. I. Spataru, C. Radocivi, and M. D. Iorga, Advances in Composite Materials: Ecodesign and Analysis, Intech, Croatia, 2011.
-
- 33. F. Asempour, D. Emadzadeh, T. Matsuura, and B. Kruczek, Desalination, 439, 179 (2018).
-
- 34. J. Guo and J. M. Catchmark, Carbohyd. Polym., 87, 1026 (2012).
-
- 35. H. J. Lee, S. K. Kim, Y. H. Kang, and W. S. Kim, J. Korean Soc. Adv. Comp. Struc., 8, 49 (2017).
-
- 36. M. L. Auad, V. S. Contos, S. Nutt, M. I. Aranguren, and M. E. Marcovich, Polym. Int., 57, 651 (2008).
-
- 37. A. Pei, J.-M. Malho, J. Ruokolainen, Q. Zhou,, and L. A. Berglund, Macromolecules, 44, 4422 (2011).
-
- 38. D. Mohan, M. S. Sajab, H. Kaco, S. B. Bakarudin, and A. M. Noor, Nanomaterials, 9, 1726 (2019).
-
- 39. J. Lyu, K. Xu, N. Zhang, C. Lu, Q. Zhang, L. Yu, F. Feng, and X. Li, Molecules, 24, 1667 (2019).
-
- 40. H. Dodiuk and S. H. Goodman, Handbook of thermoset plastics, Elsevier, San Diego, 2013.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 0379-153X(Print)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2023 Impact Factor : 0.4
- Indexed in SCIE
 This Article
This Article
-
2020; 44(3): 397-407
Published online May 25, 2020
- 10.7317/pk.2020.44.3.397
- Received on Mar 3, 2020
- Revised on Mar 23, 2020
- Accepted on Mar 25, 2020
Services
Shared
Correspondence to
- KiRyong Ha
-
Department of Chemical Engineering, Keimyung University, Daegu 42601, Korea
- E-mail: ryongi@kmu.ac.kr
- ORCID:
0000-0002-8198-7673
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr