





- Degradation Behaviors of Poly(L-lactic acid) Microspheres
Lie Ma*,**,#, Ailin Shen**,#, Jiayu Gu*, Honghua Hu**, Guoshou Jin**, Jiangfeng Cai**, Bing Feng**, Xiaodong He**,†
 , and Jun Ling*,†
, and Jun Ling*,† *MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310058, China
**Zhejiang Wedu Medical Co., Ltd., Hengdian Industrial Zone, Dongyang 322118, China- 폴리(L-락트산) 마이크로스피어의 분해 거동
Reproduction, stored in a retrieval system, or transmitted in any form of any part of this publication is permitted only by written permission from the Polymer Society of Korea.
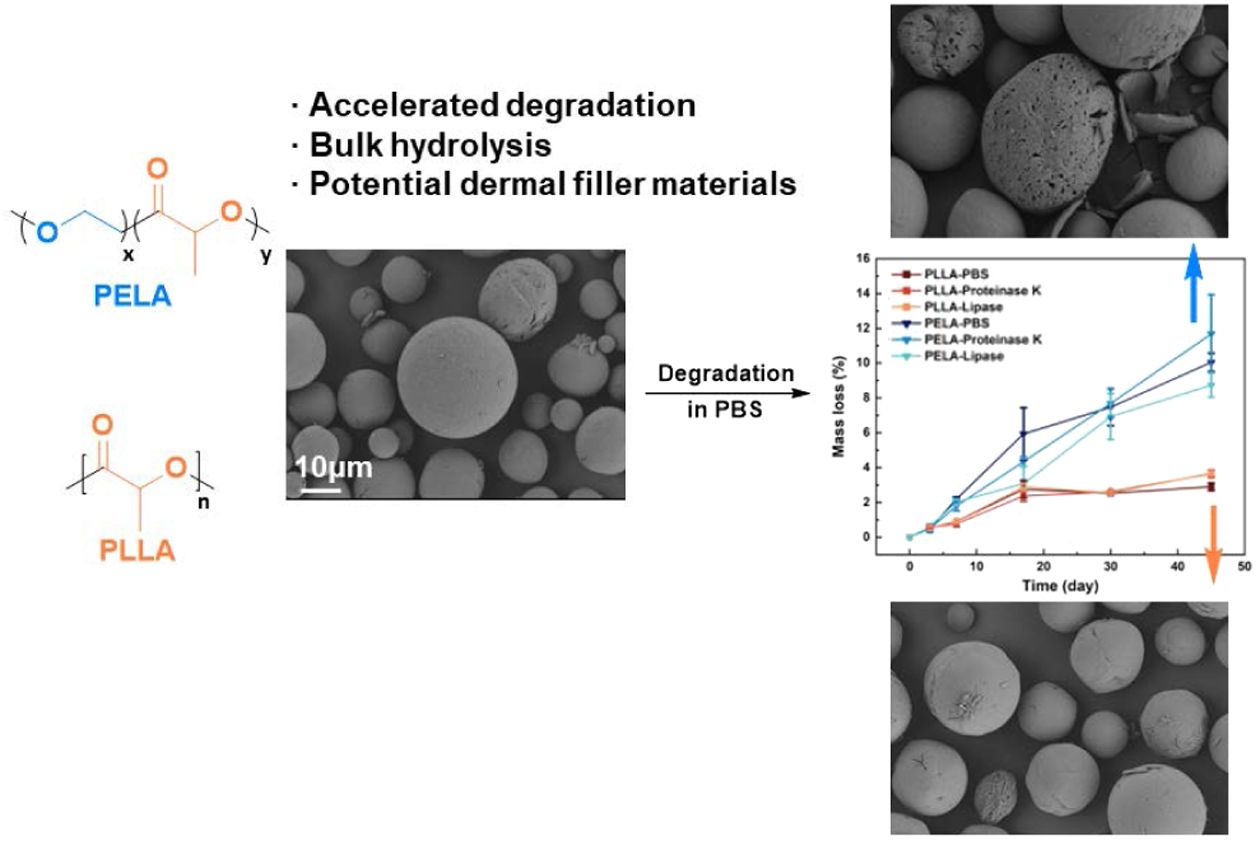
Poly(L-lactide) (PLLA) microspheres have excellent biocompatibility and biodegradability and already been applied as biomedical materials in tissue engineering. However, hydrophobic PLLA exhibits low degradation rate, bringing an obstacle to the generation of new tissues. Incorporating hydrophilic blocks endows diblock copolymer poly(L-lactide)-b-poly(ethylene glycol) (PELA) with improved biodegradability. In the present contribution, degradation behaviors of PLLA and PELA microspheres including mass loss, molecular weight changes, viscosity variation and microscopic morphology difference are monitored and analyzed. The accelerating of PEG blocks is validated. The addition of enzymes including proteinase K and lipase shows negligible effect on the degradation, thus hydrolysis in bulk polymer is proved to be the dominant degradation mechanism. Degradation characteristics including accelerates degradation rate, low surface degradation efficiency and hydrolysis nature make PELA microspheres promising candidate for dermal filler materials.
Degradation behaviors of poly(L-lactic acid) (PLLA) and poly(L-lactide)-b-poly(ethylene glycol) (PELA) microspheres were investigated under different conditions, including pure phosphate buffered saline (PBS), PBS with lipase and PBS with proteinase K. Acceleration effect of poly(ethylene glycol) (PEG) blcok was proved and the mechanism of degradation was discussed in detail.
Keywords: degradation, biomaterials, sustainable materials, block copolymers.
We thank Prof. Xufeng Ni, Dr. Peng Zhou and Mr. Yixuan Mei from Testing and Analysis Center of Department of Polymer Science and Engineering, Zhejiang University for the SEC and SEM measurements.
The authors declare that there is no conflict of interest.
Information is available regarding the detailed characterization of the microspheres. The materials are available via the Internet at http://journal. polymer-korea.or.kr.
PK_2026_050_02_220_Supporting_Information.pdf (355 kb)
Supplementary Information
Poly(lactic acid) (PLA) is one of the most commercially used biopolymers. Derived from lactides which are conveniently acquired from agricultural byproducts, PLA is considered as a suitable alternative to petrochemical-based polymers.1 PLA microspheres, known for its good biocompatibility and biodegradability, is regarded as a promising candidate for biomedical application including drug delivery,2-4 tissue engineering5-8 and dermal fillers.8 Poly(L-lactic acid) (PLLA) is one of FDA approved nonpermanent dermal filler. Different from traditional dermal fillers which take effect by volume augmentation, implanted PLLA stimulate fibroblast production, allowing cells to grow over it around the organ defect. When the healing process is finished, the biodegradable implanted fillers require for complete and safe in vivo degradation. However, application of PLLA microspheres is hindered by its slow degradation rate.9
The main degradation mechanism of PLLA is thoroughly investigated to be hydrolysis of ester bonds on the backbone.10
Under specific conditions, the cleavage of ester bonds could also be enhanced by enzymes and microorganisms.11,12
It is reported that degradative enzymes such as proteinases13 and lipases14 could localize on the surface of PLA, participating in biodegradation process. As the degradation on the surface carries on, enzymes are able to spread inside the materials thus facilitate the total degradation of PLLA.15
Due to hydrophobicity, PLLA is hard to contact water or enzymes. Complete degradation of PLA usually takes a few weeks, while the stereoregular PLLA takes up to a few months.16
To accelerate the degradation of PLLA, there are several methods including blending, copolymerization, compounding and surface modification.9
Copolymerization with hydrophilic monomers helps PLLA chains better contact with water. Poly-(L-lactide)-poly(ethylene glycol) (PELA) is a useful copolymer. Introduction of poly(ethylene glycol) (PEG) blocks endows the copolymer hydrophilicity and biocompatibility. Hydrolysis degradation of PLA could be accelerated by incorporating PEG fragments.17-20
On the other hand, PEG was reported to impede enzymatic degradation of poly(e-caprolactone) (PCL), another hydrophobic aliphatic polyester.21
The degradation rate of PLLA is dependent of its crystallinity, morphology properties and water diffusion rate into the polymers. However, there are limited reports on the degradation behavior of PELA microspheres by enzymes.
Herein, in vitro degradation behavior of PLLA and PELA microspheres is studied. Effect of PEG blocks is assessed by monitoring the mass loss, molecular weight change, particle size variation and surface morphology under incubation conditions with and without enzyme proteinase K or lipase.
Materials & Measurements. Poly(L-lactide) (PLLA, Wedu medical), Poly(L-lactide)-b-poly (ethylene glycol) (PELA, Wedu medical), KH2PO4 (AR, Sinopharm Chemical Reagent), NaOH (AR, Sinopharm Chemical Reagent), Chlorhexidine digluconate (20%, Sigma Aldrich), Proteinase K (³30 U/mg, Sigma Aldrich), Lipase type Ⅱ (125U/mg, Sigma Aldrich), dichloromethane (DCM, Shenya Chemical Co.).
A size-exclusion chromatography (SEC) composed of Agilent 1260 infinity II equipment and two CHCl3 columns (PLgel 5 μm MIXED-C 300×7.5 mm) was used to determine the molecular weights and dispersities (Đ) of polymers. CHCl3 was used as eluent with a flow rate of 1 mL/min at 35 ℃. Polystyrene was used as calibration standards. Proton nuclear magnetic resonance (1H NMR) were recorded on a JEOL JNM-ECZL400S spectrometer (1H: 400 MHz) using CDCl3 as solvent. The size of microspheres was obtained from dynamic light scattering (DLS) at 25 ℃ using Malvern MASTERSIZER 3000 particle analyzer. The morphology of microspheres was analyzed by scanning electron microscopy (SEM, ZEISS Sigma 500). The samples were sputtered with an Au/Pd coating. The coating procedure was repeated three times. Thermal properties of samples were tested by differential scanning calorimetry (DSC) (DSC Q200 TA Instruments). Thermograms were obtained under nitrogen flow (50 mL/min) at a heating and cooling rates of 10 ℃/min in the temperature range from room temperature to 200 ℃.
Preparation of Microspheres. Both PLLA and PELA microspheres were prepared according to double emulsion solvent evaporation method in CHCl2 and water.
In vitro Degradation. Preparation of Incubation Buffer: 13.6 g KH2PO4 was added to 790 mL NaOH solution (0.1 mol/L). The mixture was d iluted with water up to 2000 mL. The pH of the buffer is 7.4. 0.08 mg/mL lipase and 3.33 mg/mL proteinase K was added to the buffer, respectively. 0.5% (v/v) solution of chlorhexidine digluconate was added to each solution. 0.5 g PLLA/PELA microspheres were placed into the test tubes with 100 mL buffers (PBS/PBS with proteinase K/PBS with lipase). Then the test tubes were incubated at constant temperature of 37 ℃ in a shaking bath, set up at 120 rpm/min. To keep the enzyme effective, 1 mL newly prepared buffer was added to the degradation mixture every week.
Samples of microspheres were taken on the 3rd, 7th, 17th, 30th and 45th day since degradation.
Mass loss measurements were conducted after thoroughly isolation of samples by filtration with DI water for three times. The isolated product on the filter membrane was dried under vacuum at 40 ℃. The mass loss rate was calculated as follows:
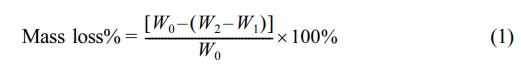
Where W0 is the weight of microspheres before incubation, W2 is the total weight of dried microspheres with filter membrane, and W1 is the weight of unused filter membrane.
After measurement of mass loss, the viscosity of samples was measured by Ubbelohde viscometer, using CHCl3 as solvent over 3 parallel samples. Molecular weights of the samples were monitored by SEC. Microscopic and the geometrical structure of the surface of the samples were recorded by SEM. Changes in the size of the microspheres were tested by DLS.
PLLA and PELA microspheres were synthesized through double emulsion solvent evaporation method. Their number average molecular weights (Mn) were comparative as 33 kg/mol and 36 kg/mol for PLLA and PELA, respectively, with the same dispersity of 1.20 determined by SEC. PELA contained 34 mol% of PEG.
Size distributions of the microspheres were characterized by DLS (Figure S1). The average diameters of microspheres were 25 μm. Analyzed by SEM in Figure 1(a) and (b) both two types of microspheres were evaluated to be spherical particles with a homogeneous size. Compared with PLLA, the surface of PELA microspheres is less smooth (Figure S2). The spongy surface could provide tunnels for small moleculeslike water to get into the microspheres freely.17
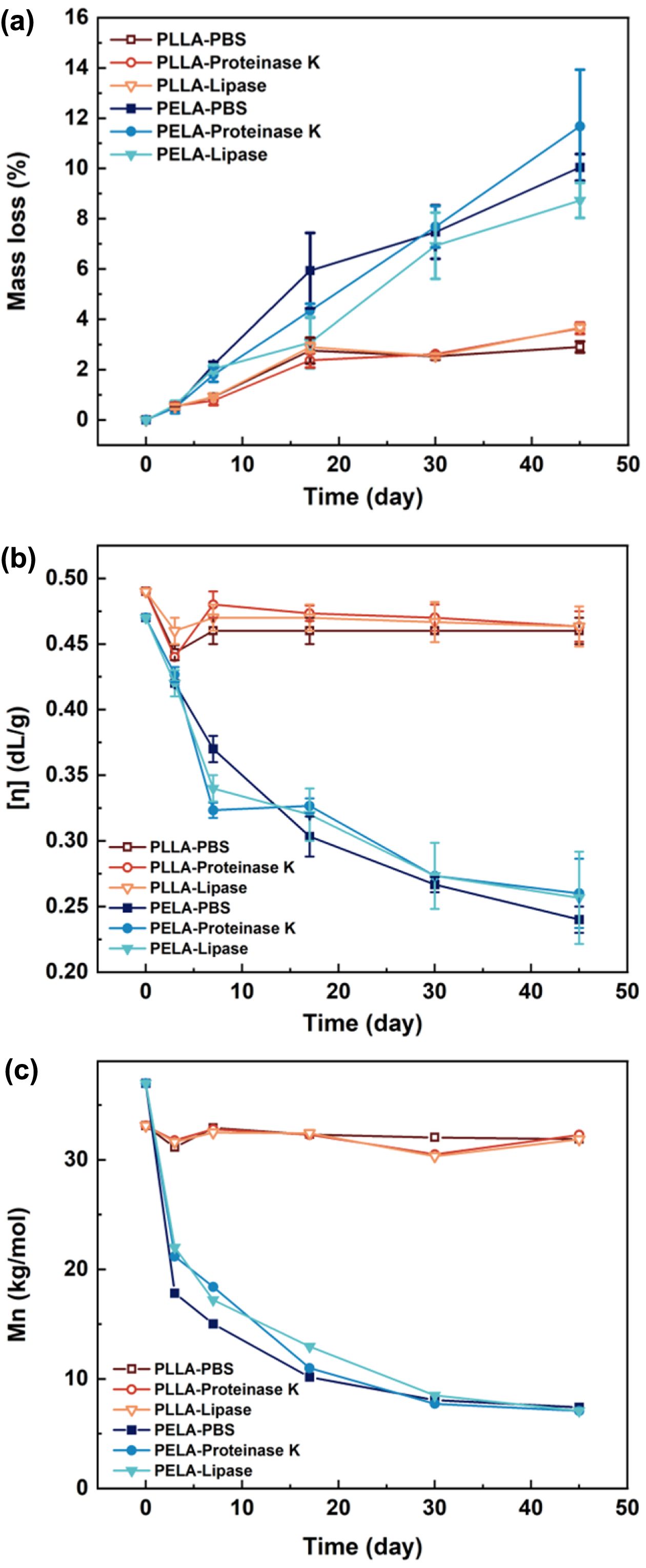
After 45 days of incubation in PBS, an average mass loss of 3% was detected in PLLA microspheres (Figure 2(a)), while the mass loss of PELA reached 10%. It indicates that hydrophilic PEG-block promotes the efficiency of chain fragmentation in microspheres. Changes in the viscosity and molecular weights agree well with the mass losses. The intrinsic viscosity of PELA continuously decreased with time from 0.45 to 0.25 (Figure 2(b)) in line with the progressive reduction of Mn from 36 kg/mol to 8 kg/mol (Figure 2(c)). Note that the degradation carried out faster within the first 20 days. In contrast, the viscosity and molecular weight of PLLA microspheres only exhibited a slight reduction. The effect of enzymes was hardly demonstrated.
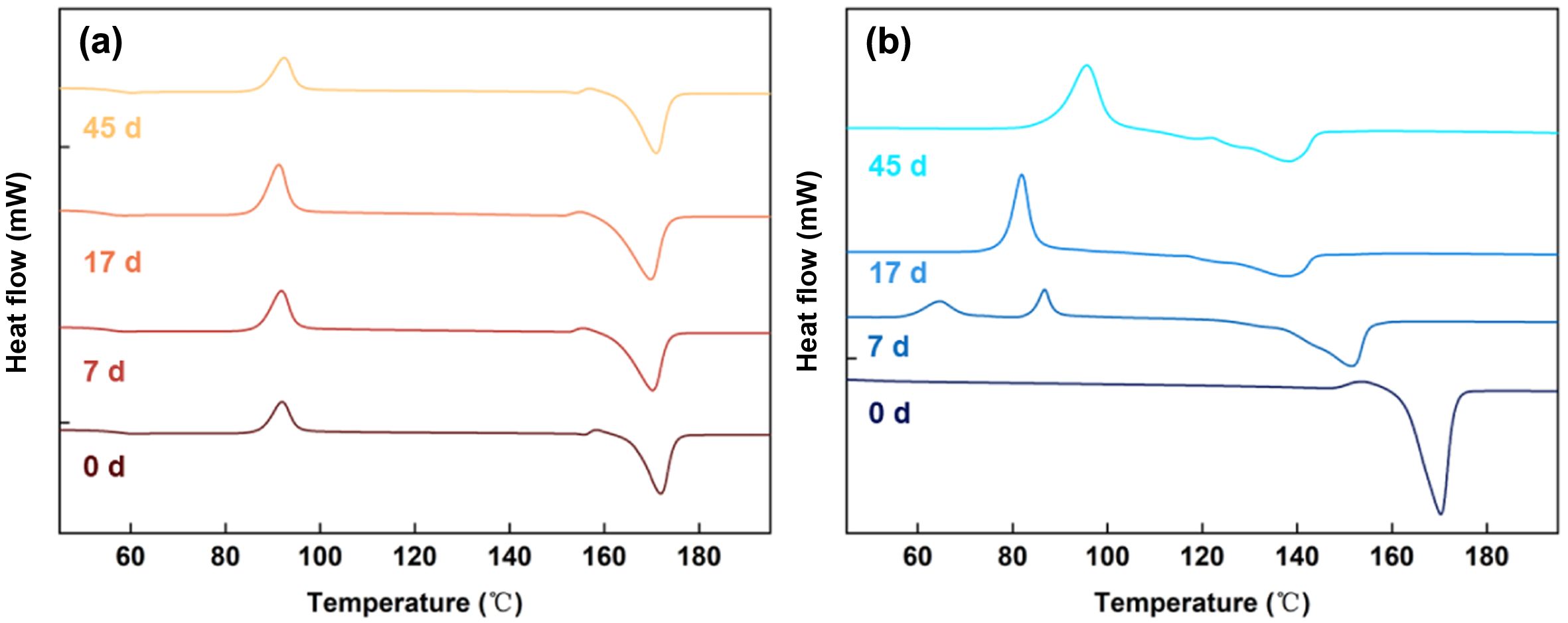
DSC was incorporated to get better knowledge of the microstructure behaviors during degradation. Before degradation (Figure 3), PLLA exhibited a glass transition at around 60 ℃ and a melting process at 175 ℃. As the Tg and Tm changed slightly after 45 days of degradation, the crystallinity of PLLA was hardly affected much by hydrolysis. The glass transition temperature was not captured for PELA, which could be ascribed to the fragment mobility enhanced by PEG block. The melting temperature of PELA decreased with degradation time in the first week, in agreement with the rapid reduction of molecular weight recorded in Figure 2. The unimodal melting peak turned into several broadened ones, indicating heterogeneity of the hydrolysis process.
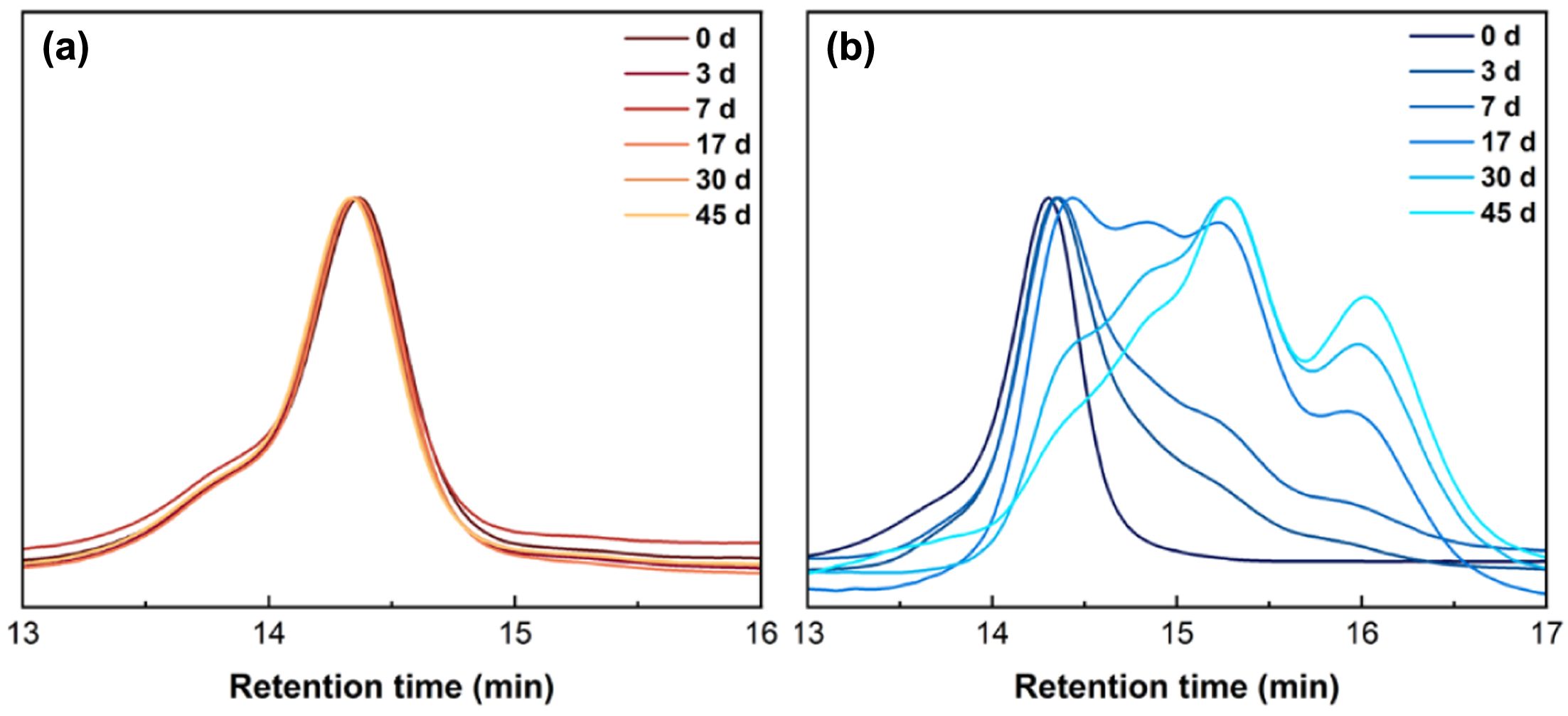
Figure 4 shows the SEC curves of microspheres incubated in PBS at different time stages. The curve of PLLA kept its unimodal peak with negligible change after 45 days of incubation. In the first week of degradation, the SEC curve of PELA shifted to the low molar mass region. At this stage, the degradation mainly happened through breakage of long polymer chains. After 17 days, multiple peaks were observed as bulk degradation had been accelerated following the mechanism proposed in literature,17
where water gradually permeated into the microspheres matrix to facilitate hydrolysis of ester bonds in bulk.
PEG content was calculated from 1H NMR spectra of the samples (Figure S3) and the results are listed in Table S2 (Supporting Information). After 45 days of degradation, PEG content decreased from 25% to 20%. The fragments connected to PEG blocks were more likely to stay in contact with water and dissolve in the buffer. As PEG is not hydrolytic under the previous condition, its content in PELA did not vary much after degradation. Rather than direct hydrolysis of itself, PEG blocks boosted the degradation process by promoting hydrolysis of PLLA through improved hydrophilicity.
The SEM images of microspheres after 45 days of incubation are shown in Figure 1(c)-(h). The freshly prepared microspheres exhibit intact surfaces, as the pores are only visible at close examination. After degradation, the sizes of both PELA and PLLA particles remain the same (Figure S1), while the sizes of pores on the surfaces of PELA microspheres increase obviously. Neither the morphology nor the pores on the surface of PLLA microspheres changes as much as PELA. The discrepancy is also attributed to the hydrophilic PEG shell which motivates the water absorption of microspheres to accelerate the hydrolysis of polymer chains near the surface. Hydrolysis and dissolution of PLLA fragments containing PEG blocks result in obvious damage on the surface of microspheres and leave sufficient tunnels for water molecules. Thus, degradation occurs both on the surface and inside the bulk microspheres. As the ether bond of PEG shows no hydrolytic reactivity, most of polymer on the surface were reserved throughout degradation. Therefore, bulk degradation mechanism is proved rather than surface one resulting in mass loss without variation in the external shape of particles. Combined with the SEC curves in Figure 4, it is demonstrated that in the first week the degradation concerning PEG blocks happened, causing pores expansion on the surface. Later on, as penetration of water into microspheres accelerated through the expanded pores, generation of oligomers was boosted, leading to growth of multi-peaks in SEC traces. It is worthy of mentioning that the micropores on the surfaces provide the possible tunnels for cell migration when the materials are used as dermal filler materials.
The proteinase K and lipase are proved to have no significant effect on the degradation. No obvious difference between the samples is detected neither on the macroscopic nor microscopic level. It was verified by several studies that enzymes including proteinase and lipase could be localized on the surface of PLA films or fibers to accelerate the degradation process.14
Materials with larger surface area adsorb enzymes in a more efficient way.22
Compared to fibers and films, however, microspheres present the smallest specific surface area, leading to weaker contact ability with enzymes. In agreement with the analysis above, the microspheres degraded only in a hydrolytic pattern. The enzyme-independent degradation is an advantage of microspheres fillers so that the individual differences among the clients would be suppressed.

|
Figure 1 The scanning electron microscope (SEM) images of (a) PLLA; (b) PELA microspheres before and after 45 day degradation: (c) PLLA in PBS; (d) PELA in PBS; (e) PLLA in PBS with proteinase K; (f) PELA in PBS with proteinase K; (g) PLLA in PBS with lipase; (h) PELA in PBS with lipase. |
|
Figure 2 (a) Mass loss; (b) the changes of intrinsic viscosity; (c) molecular weight of PLLA and PELA microspheres during degradation in PBS with and without enzymes. |
|
Figure 3 DSC thermograms of (a) PLLA; (b) PELA microspheres at different times during incubation in PBS. |
|
Figure 4 SEC traces of (a) PLLA; (b) PELA microspheres at different times during incubation in PBS. |
Introduction of PEG block efficiently promote the biodegradability of PLLA microspheres. Degradation behaviors present negligible differences from pure PBS and enzymatic conditions including proteinase K and lipase. Hydrolysis in bulk is proved to be the dominant degradation pathway. Instead of directly breaking from backbone, hydrophilic PEG fragments accelerate bulk hydrolysis of microspheres by enhancing water absorption. The bulk degradation mechanism is proved.17 All the degradation characteristics allow the PELA microspheres to be ideal dermal filler material. Hydrolytic degradation makes it possible for fillers to go through fast and safe degradation process in vivo. PELA microspheres fillers are able to keep its external shape before complete degradation, which facilitates the regeneration of collagen fibers around the microspheres.
- 1. Taib, N.-A. A. B.; Rahman, M. R.; Huda, D.; Kuok, K. K.; Hamdan, S.; Bakri, M. K. B.; Julaihi, M. R. M. B.; Khan, A. A Review on Poly Lactic Acid (PLA) as a Biodegradable Polymer. Polym. Bull. 2023, 80, 1179-1213.
-
- 2. Anderson, J. M.; Shive, M. S. Biodegradation and Biocompatibility of PLA and PLGA Microspheres. Adv. Drug Delivery Rev. 1997, 28, 5-24.
-
- 3. Qi, F.; Wu, J.; Li, H.; Ma, G. Recent Research and Development of PLGA/PLA Microspheres/nanoparticles: A Review in Scientific and Industrial Aspects. Front. Chem. Sci. Eng.2019, 13, 14-27.
-
- 4. Seol, E.; Yoon, K.; Lee, H.; Jo, S.-J.; Park, J., II; Hwang, S.-J.; Cho, C.-W. Donepezil-loaded Poly(D,L-lactic acid) Microspheres for Potent and Sustained Drug Release in the Treatment of Alzheimer’s Disease. Polym. Korea 2022, 46, 238-245.
-
- 5. Qiu, Q.-Q.; Ducheyne, P.; Ayyaswamy, P. S. New Bioactive, Degradable Composite Microspheres As Tissue Engineering Substrates. J. Biomed. Mater. Res. 2000, 52, 66-76.
-
- 6. Shi, X.; Jiang, J.; Sun, L.; Gan, Z. Hydrolysis and Biomineralization of Porous PLA Microspheres and Their Influence on Cell Growth. Colloid Surface B. 2011, 85, 73-80.
-
- 7. Lopes, M. S.; Jardini, A. L.; Filho, R. M. Poly(Lactic Acid) Production for Tissue Engineering Applications. Procedia Eng. 2012, 42, 1402-1413.
-
- 8. Ballin, A. C.; Brandt, F. S.; Cazzaniga, A. Dermal Fillers: An Update. Am. J. Clin. Dermatol. 2015, 16, 271-283.
-
- 9. McKeown, P.; Jones, M. D. The Chemical Recycling of PLA: A Review. In Sustainable Chemistry, 2020, 1, 1-22.
-
- 10. Zaaba, N. F.; Jaafar, M. A Review on Degradation Mechanisms of Polylactic Acid: Hydrolytic, Photodegradative, Microbial, and Enzymatic Degradation. Polym. Eng. Sci. 2020, 60, 2061-2075.
-
- 11. Hegyesi, N.; Zhang, Y.; Kohári, A.; Polyák, P.; Sui, X.; Pukánszky, B. Enzymatic Degradation of PLA/cellulose Nanocrystal Composites. Ind. Crop. Prod. 2019, 141, 111799.
-
- 12. Karamanlioglu, M.; Preziosi, R.; Robson, G. D. Abiotic and Biotic Environmental Degradation of the Bioplastic Polymer Poly(lactic acid): A Review. Polym. Degrad. Stabil. 2017, 137, 122-130.
-
- 13. Qi, X.; Ren, Y.; Wang, X. New Advances in the Biodegradation of Poly(lactic) Acid. Int. Biodeterior. Biodegrad. 2017, 117, 215-223.
-
- 14. Lee, S. H.; Kim, I. Y.; Song, W. S. Biodegradation of Polylactic Acid (PLA) Fibers Using Different Enzymes. Macromol. Res. 2014, 22, 657-663.
-
- 15. Satti, S. M.; Shah, A. A.; Auras, R.; Marsh, T. L. Isolation and Characterization of Bacteria Capable of Degrading Poly(lactic acid) at Ambient Temperature. Polym. Degrad. Stabil.2017, 144, 392-400.
-
- 16. Kliem, S.; Kreutzbruck, M.; Bonten, C. Review on the Biological Degradation of Polymers in Various Environments. Materials, 2020, 13, 4586.
-
- 17. Li, X.; Deng, X.; Yuan, M.; Xiong, C.; Huang, Z.; Zhang, Y.; Jia, W. In Vitro Degradation and Release Profiles of Poly-D,L-lactide-poly(ethylene glycol) Microspheres with Entrapped Proteins. J. Appl. Polym. Sci. 2000, 78, 140-148.
-
- 18. Dorati, R.; Genta, I.; Colonna, C.; Modena, T.; Pavanetto, F.; Perugini, P.; Conti, B. Investigation of the Degradation Behaviour of Poly(ethylene glycol-co-D,L-lactide) Copolymer. Polym. Degrad. Stabil. 2007, 92, 1660-1668.
-
- 19. Jh, I.; Yk, L.; Km, H. Preparation and Characterization of PEG-PLA(PLGA) Micelles for Solubilization of Pioglitazone. Polym. Korea 2008, 32, 143-149.
- 20. H, H.; Yh, C.; Sc, J.; B, L.; Ms, K.; G, K.; Hb, L. Synthesis and Characterization of Biodegradable MethoxyPoly(ethylene glycol)-Poly(ε-caprolactone-co-L-lactide) Block Copolymers. Polym. Korea 2006, 30, 28-34.
-
- 21. Peng, H.; Ling, J.; Liu, J.; Zhu, N.; Ni, X.; Shen, Z. Controlled Enzymatic Degradation of Poly(ɛ-caprolactone)-based Copolymers in the Presence of Porcine Pancreatic Lipase. Polym. Degrad. Stabil. 2010, 95, 643-650.
-
- 22. Cai, Q.; Shi, G.; Bei, J.; Wang, S. Enzymatic Degradation Behavior and Mechanism of Poly(lactide-co-glycolide) Foams by Trypsin. Biomaterials 2003, 24, 629-638.
-

- Polymer(Korea) 폴리머
- Frequency : Bimonthly(odd)
ISSN 2234-8077(Online)
Abbr. Polym. Korea - 2025 Impact Factor : 1.0
- Indexed in SCIE
 This Article
This Article
-
2026; 50(2): 220-225
Published online Mar 25, 2026
- 10.7317/pk.2026.50.2.220
- Received on Jul 23, 2025
- Revised on Oct 20, 2025
- Accepted on Nov 2, 2025
Services
- Full Text PDF
- Abstract
- ToC
- Acknowledgements
- Conflict of Interest
- Supporting Information
Introduction
Experimental
Results and Discussion
Conclusions
- References
Shared
Correspondence to
- Xiaodong He** , and Jun Ling*
-
*MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310058, China
**Zhejiang Wedu Medical Co., Ltd., Hengdian Industrial Zone, Dongyang 322118, China - E-mail: xd.he@wedumedical.com, lingjun@zju.edu.cn
- ORCID:
0009-0006-5492-2336, 0000-0002-0365-1381
Hyecheon Building(Room 601), #354, Gangnam-Daero, Gangnam-Gu, Seoul 06242, Korea
TEL : 82-2-568-3860, 561-5203, 569-3860 FAX : 82-2553-6938 E-mail: polymer@polymer.or.kr